
艾叶精油微乳的表征及稳定性研究
2020-12-23 05:29墙梦捷鲁晓翔
食品与发酵工业 2020年23期
墙梦捷,鲁晓翔
(天津商业大学 生物技术与食品科学学院,天津市食品生物技术重点实验室,天津,300134)
艾叶为菊科蒿属草本植物艾的干燥叶,含有多种营养成分包括蛋白质、膳食纤维、必需氨基酸、多不饱和脂肪酸和多种活性物质(精油、类黄酮、香豆素、有机酸和多糖)。艾叶具有抗氧化、抗菌、免疫调节、神经保护和杀虫特性[1]。艾叶精油由艾草叶、茎通过水蒸气蒸馏法、超临界CO2萃取法和石油醚萃取法提取,具有高折光率和旋光度,但具有溶解性差和气味刺鼻等缺点,给其储藏和使用带来诸多不便,限制了其食品体系中的应用[2]。
微乳是将油相和水相在表面活性剂和助表面活性剂复配的作用下,自发形成粒径为 10~100 nm的稳定均一、澄清透亮的热力学稳定体系[3]。微乳液能提高难溶药物的溶解度,对药物具有缓释控释作用,其粒径小,能促进药物有效吸收,提高药物生物利用度,对于易水解和挥发性强的药物,有很好的保护作用[4]。
微乳具有稳定性好、操作简单和生物利用度高等特点[5]。本研究以艾叶精油为原料制备艾叶精油微乳,并对水包油(O/W)型艾叶精油微乳的结构进行表征,为进一步探索艾叶精油的应用与开发提供技术和工艺参数。
1 材料与方法
1.1 材料与器材
主要试剂:艾叶精油(浓度99.99%),江西省吉水县康民本草用油提炼厂;无水乙醇,天津渤化化学有限公司;吐温- 80,天津市化学试剂供销公司;1,1-二苯基-2-三硝基苯肼(1,1-diphenyl-2-picrylhydrazyl,DPPH)试剂,成都艾科达化学试剂有限公司;试剂均为分析纯。
主要仪器设备:7890n-5975型气相色谱质谱分析仪,安捷伦有限公司;MCR 301型流变仪,安东帕商贸有限公司;Mastersizer 3000型激光粒度仪,英国马尔文仪器有限公司;79-1型磁力搅拌器,北京中兴伟业仪器有限公司;JEM-2100F型透射电子显微镜,日本电子株式会社;ME104电子天平,梅特勒-托利仪器上海有限公司;DSA 100液滴分析仪,KRUSS德国克吕士公司。
1.2 实验方法
1.2.1 微乳体系的表征
1.2.1.1 滴定法构建艾叶精油微乳液拟三元相图
准确称取一定质量吐温-80和无水乙醇于烧杯中,磁力搅拌机搅拌45 min左右,然后将上述混合表面活性剂与油相以质量比9∶1、8∶2、7∶3、6∶4、5∶5、4∶6、3∶7、2∶8、1∶9混合置于50 mL烧杯中,使用磁力搅拌器400 r/min搅拌10 min,将混合物在室温条件下静置15 min,用移液管逐渐滴加去离子水,边搅拌边滴加。反复操作直至溶液由澄清变浑浊,且状态不变。记录临界点去离子水的添加量(重复2次取平均值),计算该组分在转变点时的质量分数。以混合表面活性剂、精油和去离子水作为三相用origin制作三相图,并计算面积[6-7]。
1.2.1.2 流变法鉴定微乳构型
使用流变仪,在25 ℃下,测定不同含水质量分数的微乳液(10 %~ 90 %)在不同剪切速率下(0~300 s-1)的黏度变化,使用4 cm平板,平板和样品间距0.5 mm,取样数50,间隔6 s[8]。
1.2.1.3 动态光散射
采用激光粒度仪在室温下测定粒径尺寸、分布和zeta电位,测试光源为波长为659 nm,40 mW光泵半导体激光器,测试角度为90°,测试前将样品用0.45 μm的微孔滤膜过滤除去样品中的杂质。重复测试3次,记录测试的平均粒径[9-10]。
1.2.1.4 不同类型微乳的表面张力
按照1.2.1.1的方法配制3种类型的微乳液[m(混合活性剂)∶m(艾叶精油)=7∶3]。采用液滴分析仪测定表面张力,测3次取平均值[11]。
1.2.1.5 透射电镜观察微乳状态
使用透射电子显微镜研究O/W型微乳的形态。将铜网置于蜡板上,滴加2 μL微乳,自然风干,把质量分数2%磷钨酸(pH 7.4)滴在蜡板上,将晾干的铜网倒置于染液上,负染15 min,用蒸馏水冲洗,吸干,上电镜观察[12]。
1.2.2 艾叶精油微乳稳定性及抗氧化性研究
1.2.2.1 储藏稳定性
将微乳样品置于4、25 ℃下储藏,在第1、15、30、45天时分别测定微乳的粒径。
1.2.2.2 离心稳定性
配制含水量90%(质量分数)精油微乳,于2 000、4 000、6 000和8 000 r/min下离心30 min,观察是否分层,以去离子水为空白对照。测定离心前后波长550 nm处的吸光度[13]。透光率按公式(1)计算:
(1)
式中:T,透光率;A0,离心后的微乳吸光度;A1,离心前的微乳吸光度
1.2.2.3 自由基清除率的研究
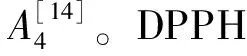
(2)
1.3 数据处理
实验均重复3次,采用Origin 8.5 对实验所得数据进行整理、图表绘制。
2 结果与分析
2.1 滴定法构建艾叶精油微乳液拟三元相图
表面活性剂和助表面活性剂的主要作用是降低互不相溶的液体之间的界面张力,在界面上形成吸附膜,促使微乳的形成[15]。拟三相图是研究微乳体系相行为变化的最简单和有效的方法,区域面积的大小反映物质载油量的多少,在制备和应用艾叶精油微乳液的过程中具有重要的指导作用[15]。由图1可知,当Km值(表面活性剂与助表面活性剂的比值)为2时,形成微乳区的面积比为53.87%。

图1 艾叶精油微乳拟三相图Fig.1 The quasi-three-phase of argyi leaf essential oil emulsion
图2分别是油包水型(W/O)、双连续相(B.C)和O/W 三种结构的实物图,分别对应含水量20%、50%和90%。该条件下配制的微乳液颜色澄清透亮,不分层不絮凝,含水质量分数为90%时微乳液呈淡蓝色乳光。

图2 含水量为20%、50%和90%的艾叶精油微乳Fig.2 Microemulsion of argyi leaf essential oil with 20%, 50% and 90% water content
2.2 流变法鉴定微乳构型
黏度是重要的物理参数,微乳液的宏观黏度会受到微观结构的影响。微乳属于牛顿型液体,采用流变仪测定不同含水量微乳液在不同剪切速率下的黏度变化[16]。艾叶精油/混合表面活性剂/水体系在Km=2时,不同含水量微乳黏度随剪切速率的变化情况如图3所示。
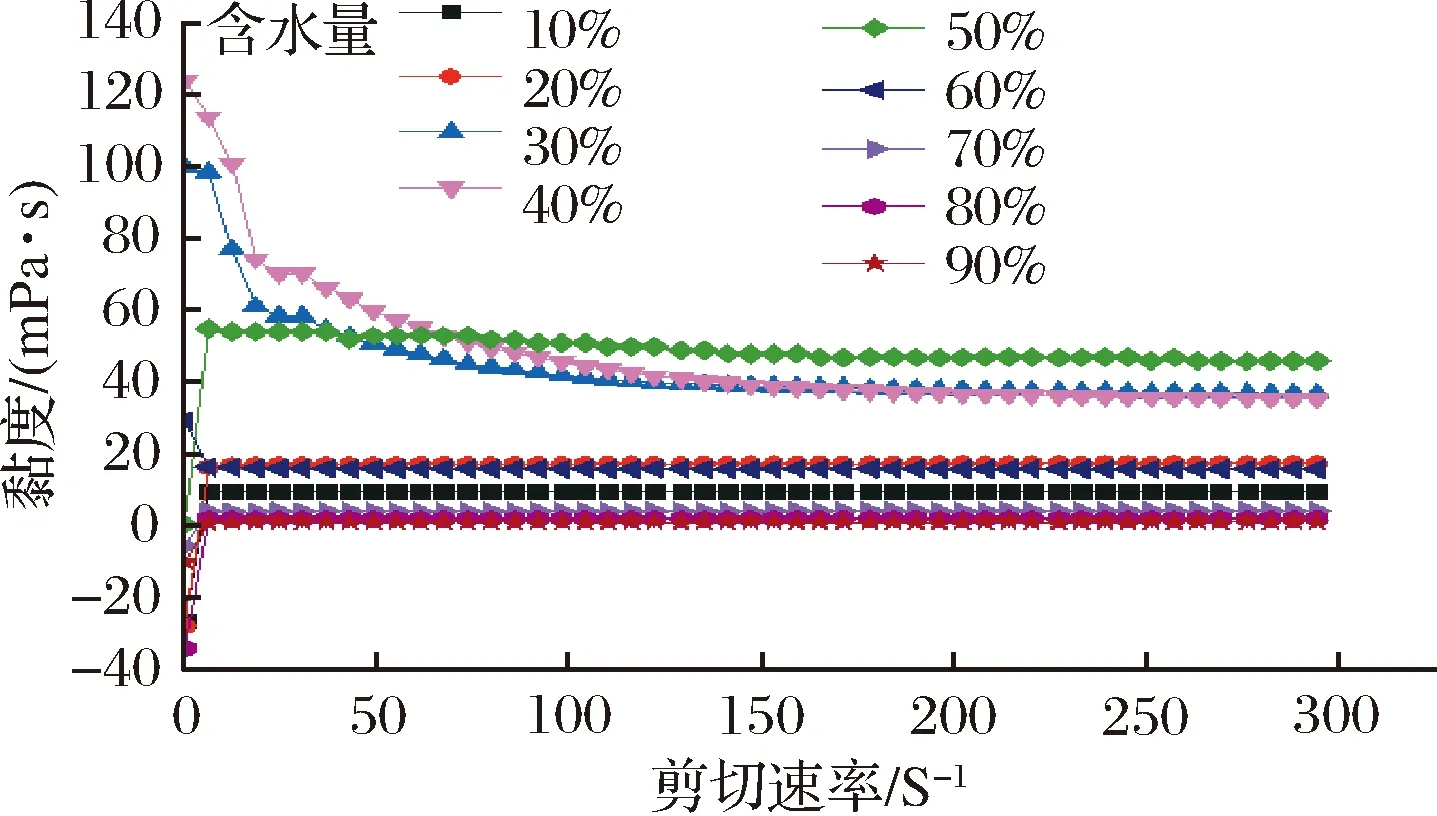
图3 不同含水量艾叶精油微乳在不同剪切速率下的黏度Fig.3 Viscosity of argyi leaf essential oil microemulsion with different water content under different shear rates
微乳构型可分为3个阶段:第1阶段,当水分含量较少时,颗粒之间的相互作用很弱,黏度接近于油,此时微乳液是W/O型;第2阶段,随着水分含量的增加,微乳液构型W/O向B.C转变,此时颗粒间作用力增大,黏度也随之上升至最大值;第3阶段,水分含量进一步增加,水作为连续相,黏度值急速下降,此时体系为O/W型结构[17-18]。为了更加直观方便的观察不同含水量微乳黏度与剪切速率的关系,选择剪切速率为153 s-1时微乳的平衡黏度来比较。当水分含量<20%时微乳的黏度在0~20 mPa·s之间变化,此时微乳的构型为W/O。水分含量从30%增加到50%时,微乳黏度增长幅度较大,这是因为此阶段双连续相构成,界面面积逐渐增加,界面之间相互作用也更加紧密,导致微乳体系黏度不断增加,含水量为50% 时达到最大黏度值(约为67 mPa·s),此时体系为B.C结构向O/W型微乳的转变点。含水量大于50% 时,体系黏度迅速降低,其原因是过量的水稀释了微乳体系,降低了胶束之间的相互作用及相互碰撞的概率[19](图4)。

图4 不同含水量艾叶精油微乳在153 s-1下的黏度Fig.4 Viscosity of argyi leaf essential oil microemulsion with different water content at 153 s-1
2.3 动态光散射
采用激光粒度仪在室温下测定艾叶精油微乳的平均粒径、zeta电位和多分散指数(polydispersity index,PDI)。由图5、图6可知,zeta电位(-2.18±0.77) mV、粒径(39.91±1.55)nm、PDI(0.28±0.068),艾叶精油制备的微乳的粒径较小,电性呈中性,体系稳定。
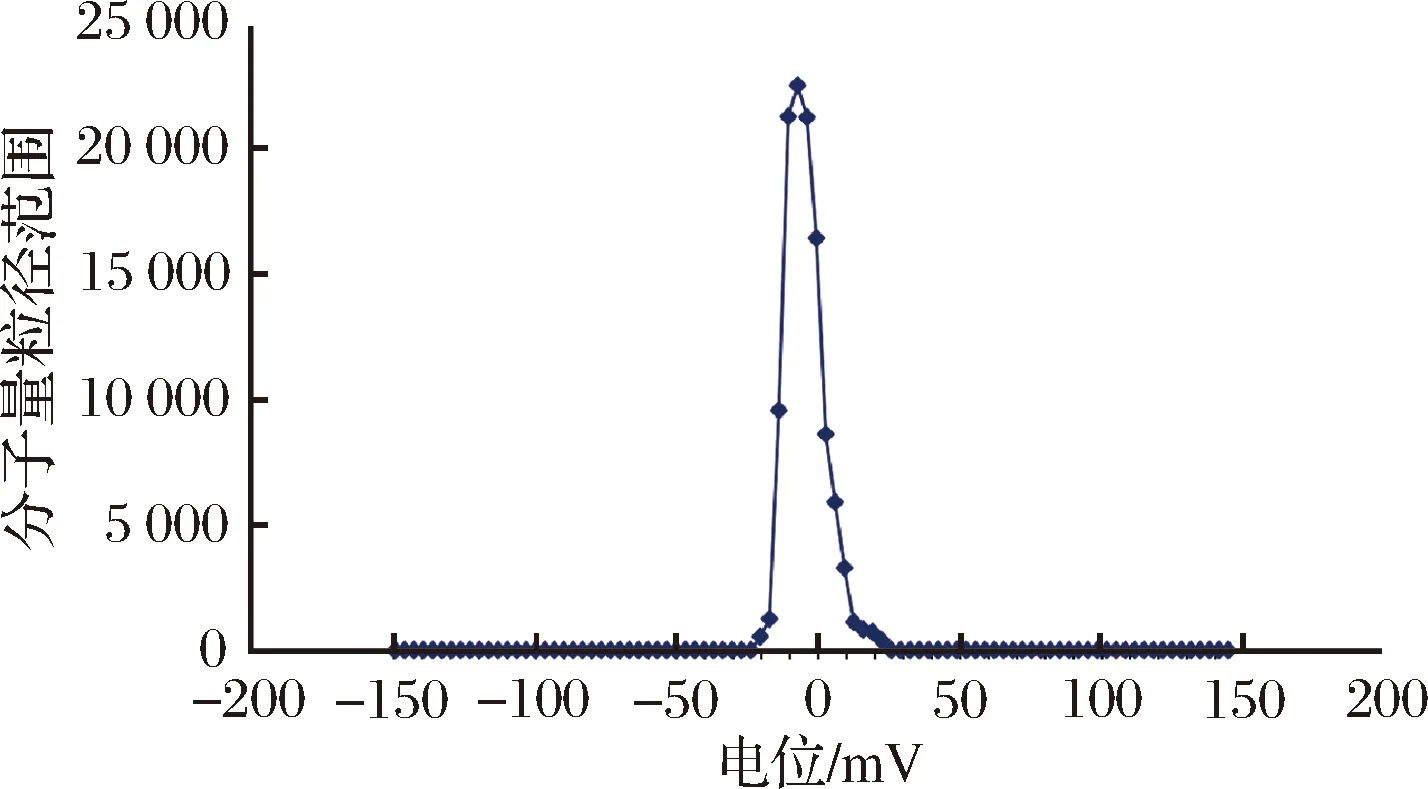
图5 O/W型艾叶精油微乳电位分布图Fig.5 Potential distribution of microemulsion of O/W argyi leaf essential oil
2.4 表面张力
采用液滴分析仪中悬滴法,当液滴在滴管口形成液滴时,此时液滴的质量和表面张力平衡。由表1可知,表面张力与温度呈负相关,即温度越高,表面张力越小。含有表面活性剂的微乳液在不同温度下表面张力的变化略大于水和乙醇,这可能是和物质结构有关。同等温度下微乳液的表面张力低于水的表面张力,这是因为表面活性剂分子与空气分子的作用力高于水分子与空气分子之间的作用力,从而导致水表面张力降低[20]。随着溶液中表面活性剂浓度的增加,排列的表面活性剂分子越多,水分子越少,溶液的表面张力降低。在20、30 ℃条件下,B.C型微乳的表面张力略大于O/W和W/O型微乳液,其原因是因为B.C相的黏度相对最大,表面活性剂迁移速度较慢,表面张力平衡时间短,容易造成水分子和表面活性剂分子不平衡[21]。

图6 O/W型艾叶精油微乳粒径分布图Fig.6 Particle size distribution of O/W type argyi leaf essential oil microemulsion
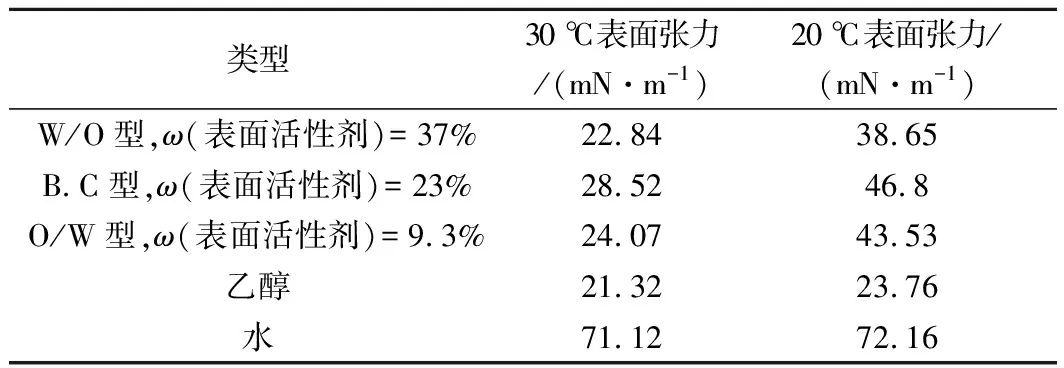
表1 不同温度下3种类型艾叶精油微乳表面张力的比较Table 1 Comparison of surface tension of different argyi leaf essential oil microemulsions at different temperatures
2.5 艾叶精油微乳透射电镜图
艾叶精油/混合表面活性剂/水体系在Km=2,稀释比为7∶3时,水分含量为90%的艾叶精油微乳在铜网上自然风干后的透射电镜图如图7所示,微乳颗粒表面呈凹凸的球状或类球状分布,该微乳体系粒子分散均匀、不聚集[22]。
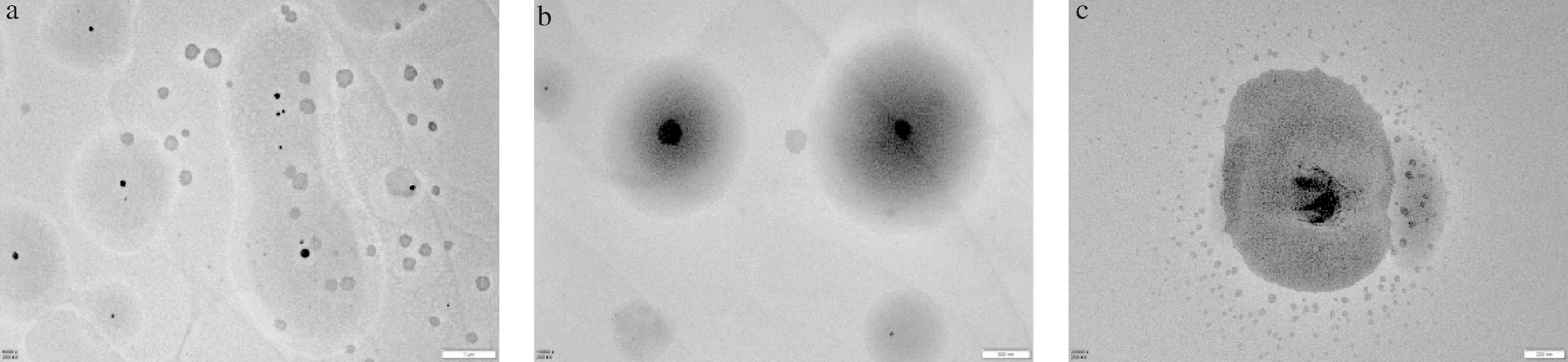
a-×6 000; b-×10 000;c-×20 000图7 O/W型艾叶精油微乳的透射电镜图Fig.7 Transmission electron microscopy of O/W type argyi leaf essential oil microemulsion
2.6 储藏稳定性
从图8可知,微乳粒径随着时间的延长而增加,在25、4 ℃条件下储藏45 d粒径分别增加了5.6和6.3 nm。不同储藏温度下,微乳粒径也会发生相应的变化,其中常温条件更有利于微乳的储藏。其原因是低温环境会降低颗粒的热能,在储藏的过程中颗粒发生少量聚集,导致体系不稳定[23]。

图8 艾叶精油微乳在不同温度下储藏时的粒径Fig.8 Particle size in argyi essential oil microemulsion stored at different temperatures
2.7 离心稳定性
为了避免微乳液在储藏、加工和运输过程中发生颗粒聚集、静置分层和破乳等现象,采用离心加速法测试微乳的稳定性,选择低、中和高3种离心速度对其进行研究。研究发现艾叶精油微乳液在中低离心速度下微乳不分层、不浑浊,其中透光率随着离心速率的增加而减少,其原因是离心力的增大使其微乳内部颗粒聚集成簇,稳定性降低[24]。

表2 艾叶精油微乳在不同离心速度下的透光率Table 2 Light transmittance of argyi leaf essential oil microemulsion at different centrifugal speeds
2.8 艾叶精油微乳自由基清除率
DPPH自由基清除率是评价物质抗氧化能力的一种简便快捷、灵敏可行的方法,并常作为检测物质抗氧化活性的指标。由图9可知,当艾叶精油微乳体系含水量<60%时,DPPH自由基清除率随着水分含量的增加而快速提高,且DPPH自由基清除率在水分含量为60% 时达到最大值。水分含量>60%时,DPPH自由基清除率随着水分含量的增加而缓慢降低。其原因是含水量较低时,体系中的水相被油相所束缚,包埋在反向胶束类,体系中多余的表面活性剂会阻碍油相(艾叶精油)与DPPH的相互接触和作用,因此随着水分含量的增加,反胶束流动性提高,油相与DPPH自由基接触面积增大,则DPPH自由基清除率快速提高[25]。当含水量>60% 时,相同的反应体积中的艾叶精油含量进一步降低,但DPPH自由基清除率的变化幅度较小,这可能是逐步增加的自由水加速了自由基反应所致,实验结果与文献[26]研究结果一致。
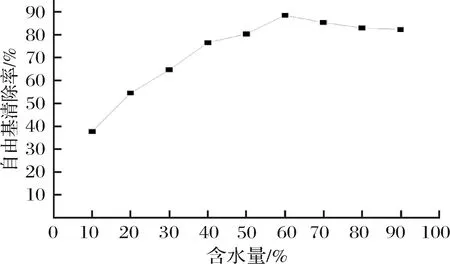
图9 不同含水量艾叶精油微乳自由基清除率的变化Fig.9 Free radical scavenging rates of argyi leaf essential oil with different water content
3 结论
本研究以艾叶精油为油相,在其表面活性剂和助表面活性剂作用下构建了一个可以提高艾叶精油利用率的四元微乳体系,研究发现艾叶精油与混合表面活性剂质量比为7∶3时,形成的微乳稳定、透亮且粒径分布在10~100 nm。为了保证该微乳在使用、运输和储藏过程中不发生质量和结构的改变,对其储藏稳定性、离心稳定性和抗氧化性进行研究。结果表明:常温条件更适合微乳的储存。通过抗氧化实验可知,O/W型微乳能呈现更好的DPPH自由基清除率。本研究研制的艾叶精油微乳剂操作简便、质量稳定,是一种天然无污染、实用低廉的新型保鲜剂,对艾叶精油微乳功能性食品制剂的开发和应用奠定了基础。
猜你喜欢
中老年保健(2022年6期)2022-08-19
上海大学学报(自然科学版)(2019年4期)2019-09-20
中成药(2017年9期)2017-12-19
中成药(2017年7期)2017-11-22
中国科技教育(2016年11期)2017-10-12
小小说月刊·下半月(2016年3期)2016-05-14
郑州大学学报(医学版)(2015年2期)2015-02-27
天然产物研究与开发(2014年8期)2014-04-27
中成药(2014年9期)2014-02-28
中国粮油学报(2013年3期)2013-09-17
