
石墨炉原子吸收法测定食品中铬
2020-12-15 09:50黄振波,李凤玉
食品安全导刊 2020年32期
铬是“中国环境优先污染黑名单”上优先监测的重金属之一,铬在环境及食物链中广泛存在和积聚,对人体呼吸道、胃肠道、皮肤等产生损伤。因此在食品风险监测中建立简便、快速、灵敏测定食品中的铬很有必要。测定铬的方法较多,主要有极谱法、原子吸收光谱法、化学发光法等,在食品风险监测中测定食品中铬最常用的方法是石墨炉原子吸收法。本文对原子吸收法测定食品中铬进行了研究,微波消解对样品进行前处理,以磷酸二氢铵作基体改进剂,直接进样检测,取得满意的结果。
1 材料与方法
1.1 仪器
1.1.1 AA240Z 型原子吸收光谱仪(附铬空心阴极灯)(VARIAN,澳大利亚)
1.1.2 微波消解仪(附聚四氟乙烯消解罐)(Anton paar,澳大利亚)
1.1.3 可调式控温电热板(Anton paar,澳大利亚)
1.2 试剂
注:除非另有规定,本方法所用试剂均为优级纯,水为GB/T 6682 规定的二级水。
1.2.1 硝酸:德国M erek 公司。
1.2.2 基体改进剂:称取2.00 g 磷酸氢二铵,加0.5 mL 硝酸,然后用水稀释定容至100 mL。
1.2.3 铬标准溶液G B W 08614 :1000 mg/mL(中国计量科学研究院);
1.2.4 铬标准使用液:用0.5%硝酸溶液逐级稀释至每毫升含100.0 ng 铬的标准使用液。
1.3 样品前处理
称取试样0.2~0.6 g(精确至0.001 g)于微波消解罐中,加入5 mL 硝酸,按照微波消解的操作步骤消解样品(条件:25℃~140 ℃,10 min、升至140℃~160 ℃,10 min、升至160℃~190 ℃,20 min)。冷却后取出消解罐,在电热板上于140℃~160 ℃赶酸至0.5 ~1.0 mL。消解罐放冷后,将消化液转移至10 mL 容量瓶中,用少量水洗涤消解罐2 ~3 次,合并洗涤液,用水定容至刻度,混匀备用。同时做试剂空白试验。
1.4 仪器测试条件
调节仪器性能至最佳状态,测定方式:峰高法,塞曼校正背景法。条件:波长357.9 nm,狭缝0.2 nm,灯电流7.0 mA。升温程序为:85 ~115 ℃干燥45 s,400 ~1100 ℃灰化17 s,2 600 ~2 750 ℃原子化3 s。
1.5 测定方法
临用时稀释铬标准溶液至铬标准使用液(20.0 μg/L),使用硝酸溶液(1%)配置6 个点以上的标准曲线:0 ~20.0 μg/L。取铬标准系列溶液10 μL、磷酸二氢铵溶液(20 g/L)5 μL,注入石墨炉中测定,测吸光值并绘制标准曲线,然后再测定样品消化液和试剂空白。
2 结果与分析
2.1 条件的选择
灰化目的是在被测元素不损失或损失极少情况下最大限度消除对被测元素的干扰,灰化温度过高可能造成被测元素损失,灵敏度降低,重复性变差;而灰化温度过低则会造成背景吸光值高。原子化是通过热分解使目标元素变成基态原子蒸气,合适的原子化温度可使被测元素得到尖锐的 吸收峰信号,并延长石墨管使用寿命。因此,分别对灰化和原子化温度进行了优化。图1 为铬元素灰化和原子化曲线图。故铬元素的最佳灰化温度为1100℃,最佳原子化温度为2600℃。基体改进剂(20 g/L 磷酸二氢铵)的加入有效提高了被测元素的热稳定性,减轻了基体干扰,吸光值明显增大。如图1所示,基体改进剂的加入量并非越多越好,当加量为5μL 时对应吸光值较高,而当加量为2和10μL 时吸光值均较加量5μL 时降低,这与文献报道的结果一致。

图1. 铬元素灰化和原子化曲线图
2.2 标准曲线性能及方法检出灵敏度
标准曲线回归方程y=0.02235x+0.00518 相关系数为0.9999,以曲线外推方程确定最佳线性范围0 ~20.0 μg/L,在上述最佳的仪器条件下,按21 次空白检测的10 倍标准差推算出方法的检出限0.15 μg/L,定量限0.50 μg/L,相对偏差1.3%。
2.3 精密度及回收实验
按本方法对3 种不同样品进行3 批×6 次测定,计算批间变异系数,表示精密度;在样品中加入低、中高3 个浓度的待测成份并分别6 次,计算回收率,表示其准确度,具体数据见表1。
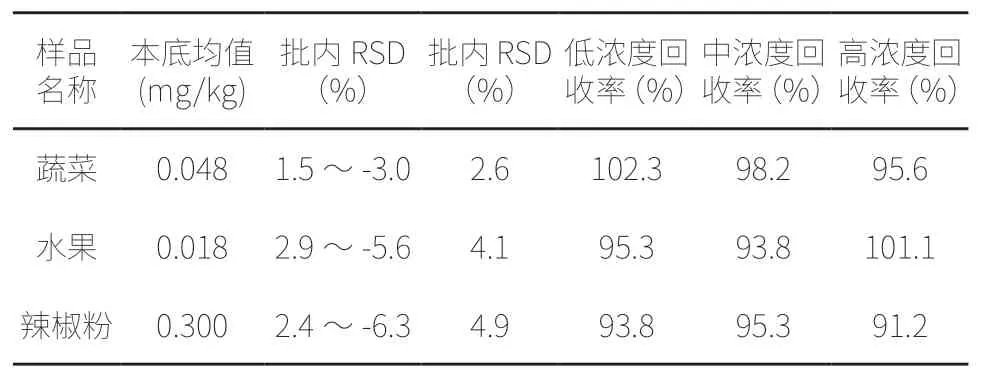
表1 方法精密度及准确度试验结果
2.4 各类食品铬的测定结果
采用微波消解法,石墨炉原子吸收标准曲线法测定广东某地区部分食品中铬含量测定结果,见表2。测定结果表明,大多数食品铬含量范围在<0.036 ~0.77 mg/kg,本方法能满足大多数食品中铬含量的检测要求。
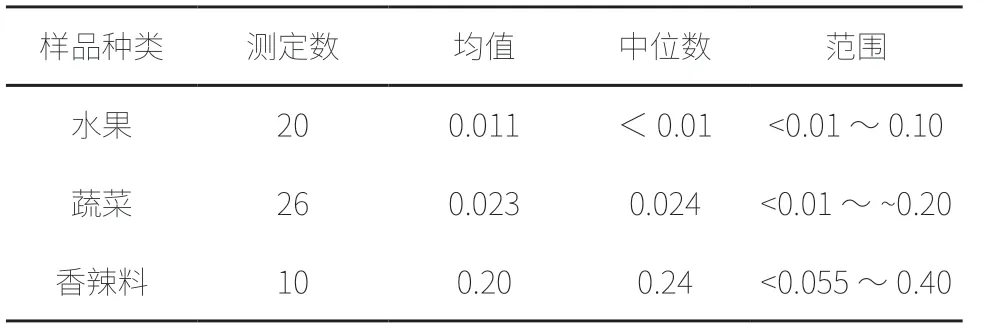
表2 广东某地区部分食品中铬含量的测定结果(mg/kg)
3 结论
采用硝酸溶液微波消解样品,选用磷酸二氢铵(20g/L)作为基体改进剂,有效去除基体干扰,使用石墨炉原子吸收法测定食品中的铬得到满意结果,该法与国标相比校,测定结果的检出限、精密度和准确度均优于国标方法,适用于食品铬的测定。
猜你喜欢
天津行政学院学报(2019年4期)2019-10-08
中国资源综合利用(2017年2期)2018-01-22
中外书摘(2017年2期)2017-02-10
广东饲料(2016年7期)2016-12-01
环境科技(2016年3期)2016-11-08
湖南有色金属(2016年3期)2016-05-18
西南农业学报(2016年4期)2016-05-17
分析测试学报(2015年7期)2016-01-13
江西理工大学学报(2015年3期)2015-12-22
社会科学研究(2014年4期)2014-08-16
