
Polyethylene glycol 35 ameliorates pancreatic inflammatory response in cerulein-induced acute pancreatitis in rats
2020-12-11 07:10:38AnaFerreroAndresArnauPaniselloRosell6JoanRosell6CatafauEmmaFolchPuy
World Journal of Gastroenterology 2020年39期
Ana Ferrero-Andres, Arnau Panisello-Rosell6, Joan Rosell6-Catafau, Emma Folch-Puy
Abstract
Key Words: Acute pancreatitis; Inflammation; Polyethylene glycols; Cytokines; AR42J cells; Cell death
INTRODUCTION
Acute pancreatitis (AP) is an inflammatory disease of the exocrine pancreas characterized by abnormal intracellular activation of proteolytic enzymes. Parenchymal injury, pancreatic acinar cell death and an intense inflammatory reaction are common pathological features of this condition and determine the severity of the disease[1]. A majority of patients presenting with AP have the mild form of the disease, which is mostly self-limited and consists of the appearance of edema and inflammation of the pancreas[2]. In this group, organ failure and local complications are generally not observed, and the disease usually resolves in the first week. However, between 20% and 30% develop a severe form requiring intensive care unit admission, which is often associated with local and systemic complications and, in some occasions, leads to death[3]. To date, no drug is available to prevent or treat this condition, and any improved clinical outcomes have mostly been due to continuous advancement of various supportive treatments.
Although pancreatic inflammation may be firstly caused by acinar events such as trypsinogen activation, it finally depends on the subsequent stimulation of components of the innate immune system. The initial acinar cell damage triggers the release of pro-inflammatory cytokines and chemokines, leading to increase of microvascular permeability and subsequent formation of interstitial edema[4]. Activation of inflammatory cells then provokes the production of additional cytokines and other mediators that initiate the inflammatory response. These mediators recruit different types of leukocytes (first neutrophils, followed by macrophages, monocytes and lymphocytes) to the pancreas. In parallel to the pro-inflammatory response, an anti-inflammatory response is also released[5]. If the anti-inflammatory response is adequate, the local inflammation resolves at this stage. However, in some cases, an overwhelming pro-inflammatory response drives the migration of inflammatory mediators into systemic circulation, leading to distant organ dysfunction[6].
Polyethylene glycols (PEGs) are hydrophilic polymers comprised of repeating ethylene glycol units[7]. PEGs have several physicochemical properties that make it advantageous in diverse biological, chemical and pharmaceutical settings, especially in view of its low toxicity. For instance, these polymers have been found to exert beneficial effects in severalin vivoandin vitromodels of cell and tissue injury[8-10].
There are very few studies linking PEGs of different molecular weight with an antiinflammatory activity. In a model of traumatic inflammation, the intraperitoneal administration of 4-kDa molecular weight PEG prevented the formation of initial adhesions and reduced the leukocytes number in the peritoneal cavity as a consequence of an inflammatory peritoneal reaction[11]. Oral treatment with 4-kDa PEG in experimental colitis reinforced the epithelial barrier function and reduced the inflammation of the colon[12]. Likewise, in two different models of gut-derived sepsis, therapeutic administration of PEG reduced inflammatory cytokine expression and activation of neutrophils[13]. Our group has recently demonstrated an antiinflammatory role for PEG35 in an experimental model of severe necrotizing AP. In this sense, the therapeutic administration of PEG35 notably alleviated the severity of AP and protected against the associated lung inflammatory response[14].
Based on the protective features of PEGs, we now have evaluated the effects of PEG35 in experimental models of pancreatic damagein vivoandin vitro.
MATERIALS AND METHODS
Experimental animals and model of cerulein-induced AP
All experimental animal proceedings were conducted according with European Union regulatory standards for experimentation with animals (Directive 2010/63/EU on the Protection of Animals Used for Scientific Purposes). The Ethical Committee for Animal Experimentation (CEEA, University of Barcelona, April 11, 2018, ethic approval number: 211/18) authorized all animal experimentation.
The protocol was designed to minimize pain and discomfort to animals. Adult male Wistar rats (n= 21) weighing 200-250 g were purchased from Charles River (Boston, MA, United States) and accommodated in a controlled environment with free access to standard laboratory pelleted formula (A04; Panlab, Barcelona, Spain) and tap water. Rats were kept in a climate-controlled environment with a 12-h light/12-h dark cycle for one week. For the 12 h prior to the experiment of AP induction, rats were fasted with free access to drinking water.
Rats were randomly selected and assigned to three equal groups: (1) Treated with saline, as controls (n= 7); and (2) Treated with cerulein, to induce AP (CerAP,n= 7) and (3) Treated with cerulein after a PEG35 pretreatment (PEG35 + CerAP,n= 7). Immediately before the first injection of PEG35 or saline, 0.05 mg/kg of buprenorphine was administered as an analgesic. Cerulein (Sigma-Aldrich, St. Louis, MO) was dissolved with phosphate-buffered saline (PBS) and administered intraperitoneally at a supramaximal stimulating concentration of 50 μg/kg/body weight (bw) at 1-h intervals (total of 5 injections); control animals received intraperitoneal saline solution with the same regime. The use of this supramaximal dosage of cerulein induce a transient form of interstitial edematous AP characterized by marked hyperamylasemia, pancreatic edema and neutrophil infiltration within the pancreas, as well as pancreatic acinar cell vacuolization and necrosis[15].
PEG35 was administered intraperitoneally at a dose of 10 mg/kg, 10 min prior to each cerulein injection. Immediately after the last injection of cerulein or saline, animals were euthanized by intraperitoneal injection of 40-60 mg/kg of sodium pentobarbital, and blood was collected from the vena cava in heparinized syringes. Harvested blood was centrifuged and the obtained plasma was stored at −80 °C until analysis. Four tissue samples from each animal were taken from the head of the pancreas. One portion of each tissue sample was immediately weighed and oven-dried for the wet-to-dry weight ratio calculation. Another portion was fixed in 10% phosphate−buffered formalin for histological analysis. The third portion was frozen and stored at −80 ºC for western blot analysis, and the last portion was saved in RNAlater solution for real-time qRT-PCR analysis.
Histopathological examination
Pancreas tissue was fixed in 10% phosphate-buffered formalin and then embedded in paraffin. 3-μm thickness sections were mounted on glass slides. Slides were dewaxed and rehydrated and stained with hematoxylin and eosin. Assessment of changes in the tissue was carried out by an experienced pathologist through the examination of different microscopic fields randomly chosen from each experimental group in a blinded manner. Pancreatic tissue sections were evaluated for the severity of pancreatitis based on edema, inflammatory infiltration, parenchymal necrosis, and vacuolation of acinar cells.
Cell lines and treatments
The rat pancreatic acinar AR42J cell line was purchased from Sigma (St. Louis, MI, United States). Cells were grown at 37 ºC in RPMI medium supplemented with 100 mL/L fetal bovine serum, 100 U/ml penicillin and 100 μg/mL streptomycin in a humidified atmosphere of 50 mL/L CO2. Acinar cells were plated at a density of 3 × 105/well in 12-well culture plates, or at a density of 2 × 104/well in 96-well plates, and allowed to attach for 24 h or 48 h. Cells were pretreated with PEG35 diluted in PBS, at a concentration of 0.5, 1, 2, 4, or 6% for 30 min prior to treatment with the appropriate stimuli: 2 µmol/L or 4 µmol/L staurosporine, 100 ng/mL TNFα or 10 nM cerulein. All three reagents were purchased from Sigma-Aldrich (St. Louis, MO, United States). Time points of 3 h were used for TNFα treatment, and of 24 h for the remaining stimuli.
Lipase activity
Plasma lipase activity levels were determined using a turbidimetric assay kit from Randox (County Antrim, Crumlin, United Kingdom), in accordance with the supplier’s specifications. Briefly, the degradation of triolein by the pancreatic lipase results in lowered turbidity, which was determined in the sample at 340 nm using a microplate reader (iEMS Reader MF; Labsystems, Helsinki, Finland). The activity of the sample was obtained in U/L. All samples were run in duplicate.
Pancreas wet-to-dry weight ratio
Edema formation in the pancreas was evaluated by the determination of the wet-todry weight ratio. A portion of the pancreas was weighed. The content of water was measured by calculating the wet-to-dry weight ratio from the initial weight (wet weight) and its weight after incubation in an oven at 60 °C for 48 h (dry weight).
Lactate dehydrogenase activity
Lactate dehydrogenase (LDH) activity was measured in plasma samples and cell culture supernatants using the Lactate Dehydrogenase Assay Kit (Abcam; Cambridge, United Kingdom). Briefly, LDH reduces NAD to NADH, which interacts with a specific probe to produce colour. Changes in absorbance due to NADH formation were measured at 450 nm at 37 °C using an automated microplate reader (iEMS Reader MF; Labsystems, Helsinki, Finland). Sample activity was expressed in mU/mL. All samples were run in duplicate. The lower limit of detection for ELISA ranged from 14 to 36 mU/mL.
MTT cell proliferation assay
The cell proliferation was determined by measuring metabolic activity of the cells through the reduction of the tetrazolium dye MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] to its insoluble formazan. AR42J cells were seeded in 96-well plates at a density of 2 × 104cells/well in 100 μL of culture medium with or without the compounds to be tested for 24 h. MTT reagent was added and incubated for 2 h at 37 °C, and the formazan produced in the cells formed dark crystals at the bottom of the wells. Crystal-dissolving solution was added and the absorbance of each sample was quantified at 570 nm using an automated microplate reader (iEMS Reader MF; Labsystems, Helsinki, Finland). All samples were run in duplicate. The absorbance intensity was proportional to the number of viable cells.
Real-time qRT-PCR
Total RNA from the pancreatic tissue and cultured cells was extracted with Nucleozol reagent (Macherey-Nagel, Dueren, Germany) in accordance with the manufacturer’s protocol. RNA concentration and quality were measured with the OD A260/A280 ratio and the OD A260/A230 ratio, respectively. Reverse transcription was performed on a 1 µg RNA sample employing the iScript cDNA Synthesis Kit (Bio-Rad Laboratories, Hercules, CA, United States). PCR amplification was performed using SsoAdvanced™ Universal SYBR®Green Supermix (Bio-Rad Laboratories, Hercules, CA, United States) on a CFX Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA, United States) using 10 µL of amplification mixture containing 50 ng of reverse-transcribed RNA and 250 nmol/L of the corresponding forward and reverse primers.
PCR primers for the detection of interleukin (IL) 6, IL1β, IL10, inducible isoform of nitric oxide synthase (iNOS), or glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were validated primers from BioRad (Hercules, CA, United States). PCR primers for tumor necrosis factor α (TNFα), designed with Primer3.0 plus[16], were: TNFα forward, 5’- ATGGGCTCCCTCTCATCAGT-3’ and reverse, 5’-GCTTG GTGGTTTGCTACGAC-3’. The specificity of amplicon was determined by melting curve analysis. Threshold cycle values were normalized to GAPDH gene expression and the ratio of the relative expression of target genes to GAPDH was calculated by the DCt formula.
(1)活动设计就是制定活动方案。活动设计考验的是学校领导的总体谋划力,基本要求是按照学校的实际情况进行长远规划、年度计划、规章制度和教育教学的实际需求,有计划、有组织地设计活动。方案是落实完成规划、计划的因子,必须与目标一致,若干个方案的计划落实最终达成目标;目标是方案的纲领,必须依靠合理的方案落实才能实现,没有方案就只能是纸上谈兵、空中楼阁。因此,活动方案既要考虑系统性、科学性,又要考虑合理性、可操作性。方案至少要讲清楚八个方面:要干什么(事由)?为什么要干(目的)?怎样干(措施)?谁来干(组织)?什么时候干(安排)?要干成什么样(目标)?干得怎样(检验)?如何干得成(保障)?
Western blot
Pancreatic tissue was homogenized in ice-cold RIPA buffer. Lysates were then centrifuged at 15000gfor 20 min at 4 °C, and the supernatants were collected. Supernatant protein concentrations were measured using the Bradford protein assay (Bio-Rad Laboratories, Hercules, CA, United States). SDS-PAGE was performed on a 10% gel and proteins were transferred to a polyvinylidene difluoride membrane for blotting.
The following antibodies were used for immunoblotting:rabbit polyclonal cleaved caspase-3 (Asp175) antibody (1:800 dilution, reference #9661) from Cell Signaling, rabbit polyclonal BCL-2 (1: 500 dilution, reference #59348) from Abcam (Cambridge, United Kingdom) and β-actin-HRP conjugated (1: 20000 dilution, reference A3854) from Sigma (Sigma-Aldrich, St. Louis, MO). Bound antibodies were detected using enhanced chemiluminescence (ECL) (Bio-Rad Laboratories, Hercules, CA, United States), and were analyzed using ChemiDoc™ Touch Imaging System (Bio-Rad Laboratories, Hercules, CA, United States). Protein expression of cleaved caspase-3 and BCL-2 were normalized to β-actin for quantification.
Statistical analysis
All data were exported into Graph Pad Prism 4 (GraphPad Software, Inc.) and presented as means ± SEM. Statistical analyses were carried out by one-way analysis of variance (ANOVA), followed by Tukey’s multiple comparison test to determine the significance between pairs. The minimal level of statistical significance was considered to be < 0.05.
RESULTS
PEG35 reduced the release of lipase associated with cerulein-induced AP
Cerulein-induced AP in rats was associated with significant raised plasma levels of lipase, comparing with the control group, reflecting the degree of pancreatic injury (Figure 1A). Such increase was significantly reduced in rats pre-treated with intravenous PEG35 at 10 mg/kg.
PEG35 abrogated pancreatic edema following cerulein-induced AP
As cerulein-induced pancreatitis is characterized by a progressive interstitial edema development, we analyzed the pancreas wet-to-dry weight ratio (Figure 1B). A significant increase in the pancreas wet-to-dry weight ratio was observed in rats after AP induction with cerulein (7.865 ± 0.86) as compared to control rats (2.76 ± 0.28). However, this increase could be largely prevented by co-treatment with PEG35 in cerulein-treated rats, with a wet-to-dry weight ratio of 3.8 ± 0.85.

Figure 1 Effect of PEG35 treatment on plasma lipase activity, pancreatic edema and histological changes in experimental ceruleininduced acute pancreatitis. A: Plasma lipase levels in U/L; B: Pancreatic wet-to-dry weight ratio. Bars represent mean values of each group ± SEM. aP < 0.05 vs control, cP < 0.05 vs Cerulein-induced acute pancreatitis (CerAP). Each determination was carried out in triplicate; C: Representative images of hematoxylin and eosin-stained pancreatic sections for each experimental group. Control group showed normal pancreas structure. CerAP group presented areas of necrosis, infiltrated polymorphonuclear neutrophils, interstitial edema and vacuolation of the acinar cells. Administration of 35-kDa polyethylene glycol notably reduced these features. Scale bar, 100 and 50 μm. CerAP: Cerulein-induced acute pancreatitis; PEG35: 35-kDa polyethylene glycol.
PEG35 reduced local pancreatic tissue damage associated with cerulein-induced AP
Histopathological results showed that cerulein hyperstimulated rats caused an interstitial edematous acute pancreatitis with considerable areas of interstitial edema, local necrosis, infiltrated polymorphonuclear neutrophils and vacuolation of the acinar cells. (Figure 1C). In the PEG35-treated group, there were consistent reductions in these characteristics.
PEG35 ameliorated the expression of inflammatory markers in cerulein-induced AP and AR42J-treated cells
Further, we explored whether PEG35 treatment improves the inflammatory response after cerulein hyperstimulation in rats, by measuring the gene expression of inflammatory mediators in the pancreas. Pancreatic tissue levels of IL6, IL1β, TNFα, IL10 and iNOS increased markedly in rats after AP induction as compared to that of control rats (Figure 2). Notably, PEG35 treatment significantly reduced the APinduced increases in IL1β, IL6 and iNOS. While TNFα expression levels showed a tendency to decrease, this was not statistically significant. Finally, as expected based on its anti-inflammatory role, the gene expression of the IL10 cytokine was not reduced in PEG35-treated animals.
In addition to itsin vivoeffects, a direct anti-inflammatory effect of PEG35 was also observed inin vitromodel. Specifically, in a model of cerulein-induced inflammation in the acinar cells using cultured AR42J cells, PEG35 attenuated the gene expression of the pro-inflammatory IL1β and TNFα in a dose-dependent manner (Figure 3A). Additionally, TNFα-treated cells induced the production of iNOS as well as of TNFα itself, both of which were markedly reduced after the treatment with increasing concentrations of PEG35 (Figure 3B).
PEG35 lessened inflammation-associated cell death in cerulein-induced AP
To investigate the potential protective effects of PEG35 on the pancreas, cell death was determined through LDH release and expression of the apoptosis-related proteins BCL-2 and cleaved-caspase-3 by Western blot. Indeed, a significant increment in LDH activity in plasma occurred in cerulein AP-induced animals (Figure 4A). Notably, rats that had PEG35 co-treatment had significantly reduced levels of the LDH necrotic marker.
The pancreatic levels of cleaved caspase-3 and BCL-2 were also markedly higher following cerulein-induced AP as compared to the control group, (Figure 4B and C). The administration of PEG35 promoted a further increase in the levels of antiapoptotic BCL-2 as compared with cerulein hyperstimulated rats while the reduction in the pro-apoptotic cleaved caspase-3 was not statistically significant.
PEG35 reduced inflammation-associated cell death in models of pancreatic damage in vitro
The decreased LDH activity observedin vivoin PEG35-treated animals led us to examine cell death inin vitromodels of inflammation. AR42J cells are a wellestablished cell model for studying intracellular mechanisms involved in the cell death and inflammatory responses of acute pancreatitis. We therefore analysed whether PEG35 affected the release of LDH in AR42J cells in the presence of the proinflammatory stimulus cerulein or TNFα (Figure 4D). Indeed, both cerulein and TNFαinduced cell death were significantly reduced by PEG35 in a dose-dependent manner. Likewise, PEG35 markedly prevented staurosporine-induced AR42J apoptotic cell death in a dose-dependent manner (Figure 4E). These results suggest that PEG35 exerts a protective role against inflammation-induced cell deathin vitroandin vivo.
DISCUSSION
Acute pancreatitis (AP) is an inflammatory disease that can have a mild to severe course. We have recently reported an anti-inflammatory role for PEG35 in a severe necrotizing AP experimental model. To further investigate the effect of this polymer in a milder form of the disease, we used a model of cerulein-induced mild edematous pancreatitis that is mainly characterized by a dysregulation of the production and secretion of digestive enzymes, interstitial edema formation, infiltration of neutrophil and mononuclear cells within the pancreas, cytoplasmic vacuolization and the death of acinar cells[17]. We determined that PEG35 reduced the course of cerulein-induced AP by inhibiting the inflammatory response as well as inflammation-induced cell death. In our study, treating the animals with PEG35 significantly abrogated the severity of cerulein-induced AP, as indicated by the lessened activity of lipase in plasma and edema formation as well as histopathological features of AP in the PEG35-treated animals.
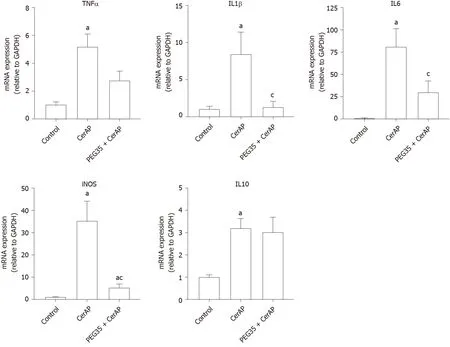
Figure 2 Role of PEG35 on the modulation of inflammation-associated cytokines and inducible nitric oxide synthase enzyme expression in cerulein-induced acute pancreatitis. Pancreatic tissue gene expression of tumor necrosis factor α, interleukin (IL) 1β, IL6, inducible nitric oxide synthase and IL10 by real-time qRT-PCR. Bars represent mean values of each group ± SEM. aP < 0.05 vs control, cP < 0.05 vs cerulein-induced acute pancreatitis. Each determination was carried out in triplicate. CerAP: Cerulein-induced acute pancreatitis; PEG35: 35-kDa polyethylene glycol.
A sudden inflammatory response in the pancreas contributes to the development of AP, primarily through the release of inflammatory cytokines. TNFα has long been considered as one of the initial triggers of the inflammatory cascade in experimental pancreatitis[18]. In this setting, stimulation of acinar cells of the pancreas by TNFα have been reported to cause a direct activation of pancreatic enzymes, contributing to premature protease activation and cell necrosis[19]. Increased accumulation of TNFα promotes the production of other inflammatory cytokines, including IL1β and IL6, which result in the activation of an inflammatory cascade that leads to widespread tissue damage in multiple tissues and organs. Indeed, the levels of TNFα, IL1β and IL6 have been correlated with the severity of AP[20-23]. In the current study, treatment with PEG35 was capable to significantly reduce the AP-induced raises in pro-inflammatory IL1β and IL6. However, no significant effect on TNFα was observed. This fact could be explained by the levels of IL10 found in rats co-treated with cerulein and PEG35, which were similar to those found in those only treated with cerulein. As IL10 plays a fundamental role in the attenuation of the cytokine response during acute inflammation, the significant increase of IL10 found in hyperstimulated rats may contribute to slow TNFα production, with an observed tendency towards a decrease in its expression. Indeed, in an experimental model of cerulein-induced AP, intraperitoneal IL10 administration attenuated TNFα production, which was associated with dramatically lessened pancreatitis severity and mortality[24].
Furthermore, a direct anti-inflammatory effect of PEG35 was observed in cultured AR42J cells. In anin vitromodel of cerulein-induced inflammation, PEG35 was able to attenuate the gene expression of pro-inflammatory IL1β and TNFα in a dosedependent manner. Moreover, PEG35 reduced the levels of TNFα in AR42J cells stimulated with TNFα.

Figure 3 Gene expressions of inflammatory markers in AR42J-treated cells. A: Gene expression by real-time qRT-PCR of tumor necrosis factor α (TNFα) and IL1β in cerulein-treated AR42J cells subjected to increasing concentrations of 35-kDa polyethylene glycol (PEG35); B: Gene expression by real-time qRT-PCR of TNFα and inducible nitric oxide synthase in TNFα-treated AR42J cells subjected to increasing concentrations of PEG35. In both cases, mRNA induction levels were normalized to GAPDH mRNA expression. Bars represent mean values of each group ± SEM. aP < 0.05 vs control, cP < 0.05 vs cerulein or TNFα. Each determination was carried out in triplicate. Cer: Cerulein; PEG35: 35-kDa polyethylene glycol; TNFα: Tumor necrosis factor α.
Pro-inflammatory cytokines are known to activate the inducible iNOS and the subsequent production of nitric oxide, thus contributing to the pathophysiology of AP. In fact, the degree of pancreatic inflammation and tissue injury of cerulein-induced AP has been found to be markedly reduced in iNOS-deficient mice[25]. In our study, we observed an increased mRNA expression of iNOS following cerulein hyperstimulation in rats, which underwent a significant reduction after PEG35 treatment. Likewise, PEG35 abrogated TNFα-induced iNOS expression in acinar cells in a concentrationdependent manner. Altogether, these results suggest that PEG35 treatment reduced pancreatic inflammation in pancreatitis by suppressing the expression of proinflammatory mediators.
These changes in the inflammatory response brought about by PEG35 treatment were further emphasized by a reduction in pancreatic cell death. PEG35 treatment reduced cell death in cerulein-induced AP rats by lowering plasmatic LDH activity. In addition, the increased release of LDH observed in cerulein and TNFα-treated acinar cellsin vitrowas reverted upon incubation with increasing concentrations of PEG35.
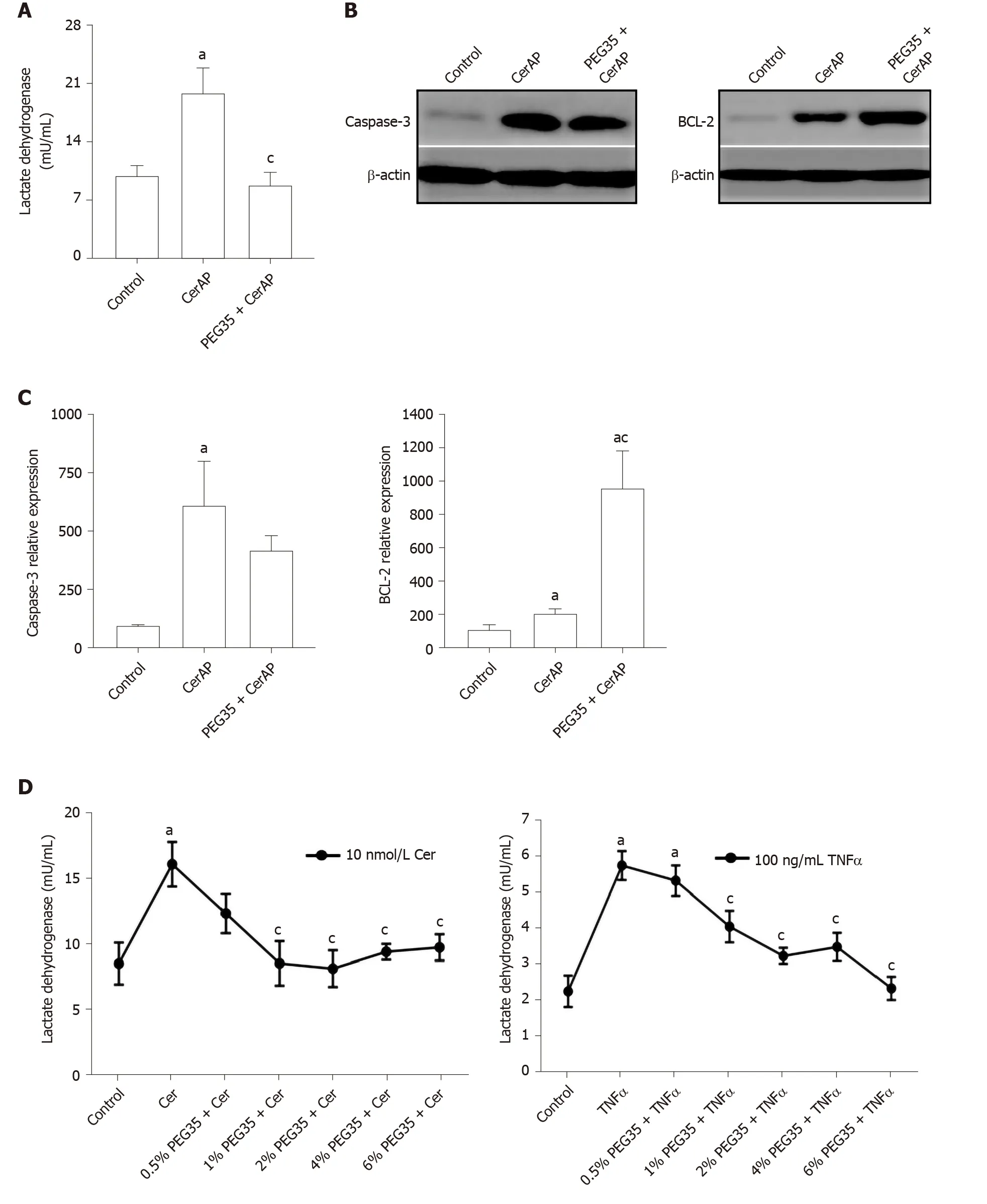
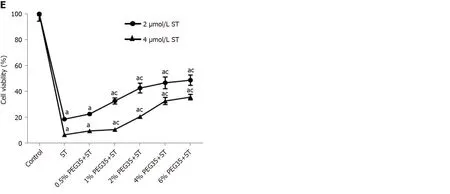
Figure 4 Effect of 35-kDa polyethylene glycol on inflammation-induced cell death in cerulein-induced acute pancreatitis and cultured pancreatic acinar AR42J cells. A: Plasma lactate dehydrogenase (LDH) activity after cerulein-induced acute pancreatitis expressed as mU/mL; B: Pancreatic protein expression of cleaved caspase-3 and BCL-2 assessed by western blot analysis. β-actin expression was used as loading control. Data shown are representative blots for each group; C: Densitometry quantification of western blot for cleaved caspase-3 and BCL-2 in pancreatic tissue; D: Cell death rate measured through LDH activity. AR42J cells pre-treated with increasing concentrations of PEG35 (0.5%, 1%, 2%, 4% or 6%) for 30 min and then co-incubated with 10nM cerulein for another 24 h or 100 ng/mL of tumor necrosis factor α (TNFα) for another 2.5 h; E: Cell viability rate determined by MTT assay. AR42J cells were pretreated with increasing concentrations of PEG35, as indicated, for 30 min and then incubated with or without 2 µM or 4 µM staurosporine for another 24 h. The values shown represent the mean ± SEM. aP < 0.05 vs control, cP < 0.05 vs cerulein-induced acute pancreatitis, cerulein, TNFα or staurosporine. Each determination was carried out in triplicate. CerAP: Cerulein-induced acute pancreatitis; Cer: Cerulein; PEG35: 35-kDa polyethylene glycol; TNFα: Tumor necrosis factor α; ST: Staurosporine.
In the pancreas, inflammation is associated with injured acinar cells that can go through necrosis or apoptosis. Thus, we measured the apoptosis index in pancreatic tissue following cerulein-induced AP. Injured pancreatic tissue induced a significant increase in cleaved caspase-3 and BCL-2 apoptotic proteins as compared to the respective controls. Following treatment with PEG35, anti-apoptotic BCL-2 further increased as compared with cerulein-treated animals while cleaved caspase-3 levels were similar to that found in cerulein hyperstimulated animals. Collectively, these findings suggest that PEG35 has anti-apoptotic and anti-necrotic properties for cerulein-induced pancreatitis.
CONCLUSION
In conclusion, results from this study reveal a mechanism by which PEG35 exerts antiinflammatory effects that alleviate experimental cerulein-induced AP, by inhibiting the inflammatory response as well as inflammation-induced cell death. Because of its low toxicity as well as its proven biocompatibility, PEG35 could be used as a new therapeutic tool to resolve the cellular damage associated to mild AP.
ARTICLE HIGHLIGHTS

Research objectives
To evaluate the effect of PEG35 in experimental models of mild acute pancreatitisin vivoandin vitro.
Research methods
AP was induced by five hourly intraperitoneal injections of cerulein (50 μg/kg/bw). PEG35 was administered intraperitoneally 10 minutes before each cerulein injection in a dose of 10 mg/kg. After AP induction, samples of pancreatic tissue and blood were collected for analysis. AR42J pancreatic acinar cells were treated with increasing concentrations of PEG35 prior to exposure with tumor necrosis factor α, staurosporine or cerulein. The severity of AP was determined on the basis of plasma levels of lipase, lactate dehydrogenase activity, pancreatic edema and histological changes. To evaluate the extent of the inflammatory response, the gene expression of inflammation-associated markers was determined in the pancreas and in AR42Jtreated cells. Inflammation-induced cell death was also measured in bothin vivoandin vitromodels of pancreatic damage through apoptosis and necrosis-related assays.
Research results
PEG35 treatment significantly improved pancreatic damage in cerulein-induced AP in rats through reduction on lipase levels and tissue edema. Furthermore, PEG35 ameliorated the inflammatory response and associated cell deathin vivoandin vitro, in treated-acinar cells, by lowering inflammatory-related cytokines and iNOS gene expression, levels of apoptotic markers and the activity of lactate dehydrogenase.
Research conclusions
PEG35 ameliorated pancreatic damage in cerulein-induced AP and cultured acinar AR42J-treated cells through the attenuation of the inflammatory response and associated cell death.
Research perspectives
Our study provided evidence of a protective role of PEG35 in a mild form of AP suggesting that PEG35 may be a valuable option in the management of clinical AP.
ACKNOWLEDGEMENTS
This study was supported by grant from Ministerio de Ciencia e Innovación (PID2019-104130RB-I00) to Emma Folch-Puy. The authors thank Veronica Raker for revising the English text.
猜你喜欢
小猕猴学习画刊(2023年3期)2023-03-27 08:56:42
英语文摘(2022年8期)2022-09-02 02:00:06
宝藏(2020年3期)2020-10-14 09:41:26
快乐语文(2020年11期)2020-06-06 04:16:36
幽默大师(漫话国学)(2018年6期)2018-11-06 07:17:02
中外文摘(2018年8期)2018-03-27 08:34:31
小学阅读指南·低年级版(2016年5期)2016-05-14 14:05:06
语文知识(2014年4期)2014-02-28 21:59:43
上海节能(2011年6期)2011-08-15 00:45:06
中国土地科学(2011年7期)2011-03-20 16:26:20
 World Journal of Gastroenterology2020年39期
World Journal of Gastroenterology2020年39期
- World Journal of Gastroenterology的其它文章
- Herbal cake-partitioned moxibustion inhibits colonic autophagy in Crohn’s disease via signaling involving distinct classes of phosphatidylinositol 3-kinases
- Identification of differentially expressed genes in ulcerative colitis and verification in a colitis mouse model by bioinformatics analyses
- Artificial intelligence technique in detection of early esophageal cancer
- Impact of cap-assisted colonoscopy during transendoscopic enteral tubing: A randomized controlled trial
- Single access laparoscopic total colectomy for severe refractory ulcerative colitis
- Acute gastrointestinal injury in critically ill patients with COVID-19 in Wuhan, China