
基于高灵敏度荧光衍生剂的痕量维生素C液相检测方法建立
2020-12-10 03:21沈海波张连钢周鑫魁方剑儒刘善鑫莫慧娟张婧蕾
食品工业科技 2020年23期
沈海波,张连钢,周鑫魁,方剑儒,洪 霞,刘善鑫,莫慧娟,张婧蕾
(1. 嘉峪关市食品药品和医疗器械检验检测中心,甘肃嘉峪关 735100;2.甘肃中商食品质量检验检测有限公司,甘肃兰州 730010;3.张掖市药品检验检测中心,甘肃张掖 734000)

现有方法中,无法同时满足灵敏度高、噪声小、操作简单的实验要求,因此建立一种操作相对简便的样品处理方法和一种高灵敏度,检测结果稳定,自动化程度高的检测方法是很有必要的,在已经公开发表的文献中尚未报道有如何利用荧光衍生-高效液相色谱法测定痕量维生素C的方法,所以本文将高灵敏度的荧光衍生方法与高效快速的液相色谱法相结合,建立一套灵敏度高、操作相对简便、稳定的食品中痕量维生素C测定方法。
1 材料与方法
1.1 材料与仪器
维生素C标准品 标准品(97.5%),德国Dr. Ehrenstorfer公司;6-羟基-2,4,5-三氨基嘧啶 优级纯,上海麦克林生化科技有限公司;草酸 分析纯,上海中秦化学试剂有限公司;乙酸钠 分析纯,上海中秦化学试剂有限公司;新鲜水果蔬菜样品 均购于超市及农贸市场。
20A高效液相色谱仪 岛津企业管理中国有限公司;XB220电子天平 普利赛斯国际贸易(上海)有限公司;RX-II型离心机 天美中国科学仪器有限公司。
1.2 实验方法
1.2.1 样品前处理方法 称取5.00 g粉碎或匀浆后的样品(包括蔬菜水果及其制品、乳及乳制品、蛋类、肉类、菌类、水产品),于50 mL离心管中,加入提取试剂1%草酸水溶液定容至50 mL,涡旋提取2 min,超声提取5 min;将上述离心管于8000 r/min离心3 min,取上清液备用。
1.2.2 衍生方法 取上清液5 mL于15 mL离心管中,用饱和乙酸钠调节pH为6.0~7.0,加入1 mL衍生试剂,40 ℃避光反应0.5 h。取清液过滤孔直径为0.22 μm的滤膜,高效液相色谱仪测定。
1.2.3 色谱条件及标准曲线绘制 色谱柱:C18柱(5 μm,4.6 mm×150 mm);流动相:pH=4.0乙酸铵缓冲液(v)+乙腈(v)+甲醇(v)=80+15+5,等度洗脱;流速:1 mL/min;柱温:35 ℃;进样量:20 μL;激发波长:373 nm,发射波长:442 nm。
配制七个不同质量体积浓度的维生素C标准溶液,质量体积浓度分别为:0.05、0.10、0.50、2.50、10.0、50.0、500.0 μg/mL;将维生素C标准溶液按1.2.2处理,按1.2.3色谱条件进入附带荧光检测器的高效液相色谱仪中测定,绘制标准曲线,并计算样品中维生素C的含量。
1.2.4 样品提取溶剂选择 样品中维生素C提取方式以溶液浸提法为主,根据文献[4,6-7,15]提取液分别为1%草酸水溶液、2%偏磷酸水溶液、1%盐酸水溶液、二次蒸馏水。分别以四种溶液为样品提取溶剂,分别按照1.2.1和1.2.2方法步骤处理标识值为322 μg/g的乳粉样品。根据检测结果及实验现象,比较四种提取溶剂。
1.2.5 衍生试剂的选择 检测痕量维生素C时常用荧光分光光度法,其衍生剂为邻苯二胺水溶液。分别使用邻苯二胺和6-羟基-2,4,5-三氨基嘧啶为衍生试剂。参考国标GB5413.18-2010方法[9]对标识值322 μg/g的乳粉样品进行衍生并检测其中维生素C的含量。
1.2.6 衍生最佳pH范围确定 按照1.2.2方法,用饱和乙酸钠及氢氧化钠溶液调节5 mL的100 ng/mL标准溶液pH分别为2.0、3.0、4.0、5.0、6.0、7.0、8.0、9.0、10.0,分别加入等量衍生液,用超纯水定容至10 mL,于40 ℃衍生0.5 h。在1.2.3色谱条件下测定不同pH下衍生液中维生素C含量。
1.2.7 衍生温度及衍生时间研究 按1.2.1方法处理维生素C含量标识值为49.2 μg/g的阳性乳粉样品,在pH为6.5±0.5条件下,参考文献[10]中的方法,同时进行衍生温度及衍生时间的棋盘实验,每种条件下做三组平行试验,并按照1.2.3色谱条件检测每种衍生条件的维生素C含量。

表1 不同提取液处理样品对比结果
1.2.8 衍生试剂浓度研究 称取一定量6-羟基-2,4,5-三氨基嘧啶,配制成0.05、0.1、0.5、1.0、2.0、5.0 g/L不同浓度的6-羟基-2,4,5-三氨基嘧啶水溶液。以六种浓度溶液,分别为衍生剂,在最佳衍生条件下对50 ng/mL的维生素C标准溶液进行衍生反应,并测定衍生产物含量。
1.2.9 方法检出限 按照1.2.3色谱条件检测空白样品,得到30 min空白样品基线色谱图,以30 min内最大基线噪声的三倍为仪器最低的可识别信号,作为仪器检出限。根据方法稀释倍数计算方法检出限。
1.2.10 维生素C含量的计算 根据1.2.2测得的样品制备液的峰面积值,从1.2.3绘制的系列维生素C标准溶液的工作曲线上找到峰面积值对应的维生素C的含量,按下式计算待测食品中维生素C的含量X:
式中:X表示试样中的维生素C含量,单位μg/g;ρ表示1.2.3步骤由标准曲线测得的样品制备液的峰面积所对应的维生素C的含量,单位μg/mL;m表示试样质量,单位g;V表示1.2.1步骤中定容体积,单位mL;f表示稀释倍数,本实验中为1;1000表示单位μg/g在适用情况下,可以转化为mg/kg的系数,可以不使用。
1.3 数据处理
本实验用相关分析,对应分析,回归分析,处理分析数据,得到相关结论。用Excel软件绘制图表,使数据结果更直观。
2 结果与讨论
2.1 样品提取液选择
四种提取液处理同一乳粉样品中维生素C含量检测结果如表1所示。
由表1可知,由于二次蒸馏水无法有效沉淀蛋白质,二次蒸馏水在提取蛋类、乳及乳制品类样品时,通过离心和过滤操作无法获得上清液;盐酸在溶液中无法与Fe3+、Cu2+形成络合物,所以1%盐酸水溶液提取含Fe3+、Cu2+较多的样品时,Fe3+、Cu2+可以结合维生素C形成络合物[21],导致所测维生素C含量偏低;以偏磷酸和草酸提取效果最佳,而偏磷酸不易溶于水,在实际操作中,配制偏磷酸溶液耗时长;为更快速、有效的测定食品中维生素C的含量,本实验采用1%草酸水溶液做样品提取液。
2.2 衍生试剂对比
邻苯二胺衍生产物为苯并含氧六元杂环。而本方法荧光衍生剂采用6-羟基-2,4,5-三氨基嘧啶,衍生产物为嘧啶并含氧六元杂环,且在嘧啶环上有邻对位的氨基和羟基,氨基和羟基为给电子基团,能使得嘧啶并含氧六元杂环的电子共轭区域增大,大幅增加衍生产物的荧光强度。在液相色谱中得到更强的荧光信号,从而有利于提高方法的灵敏度,更适合用于痕量维生素C的检测工作中。分别在两种不同的衍生剂最佳衍生条件下,充分衍生同浓度的维生素C,并测定其荧光强度,对比目标物色谱峰面积,维生素C浓度相同的条件下,色谱峰面积越大,证明其荧光强度越强。两种衍生剂所得到的色谱图如图1所示。结果表明,6-羟基-2,4,5-三氨基嘧啶的衍生产物的荧光强度是邻苯二胺的衍生产物的1.83倍。

图1 两种衍生剂衍生产物的色谱图
2.3 衍生最佳pH范围确定
以衍生pH为横坐标,衍生产物液相色谱峰面积为纵坐标,得到该曲线。如图2所示。

图2 不同pH环境中衍生效果对比
由图2可知,pH环境对衍生反应影响很大,偏酸或偏碱性溶液都不利于羰基与氨基的缩合反应,阻碍衍生进行。所以选择维生素C与衍生剂反应的最佳pH环境为6~7之间,实验选择在pH为6.5±0.5范围进行衍生反应。
2.4 最佳衍生温度及衍生时间
每组试验得到三组数据,对比结果,选出最佳衍生温度及衍生时间。条件及结果如表2所示。
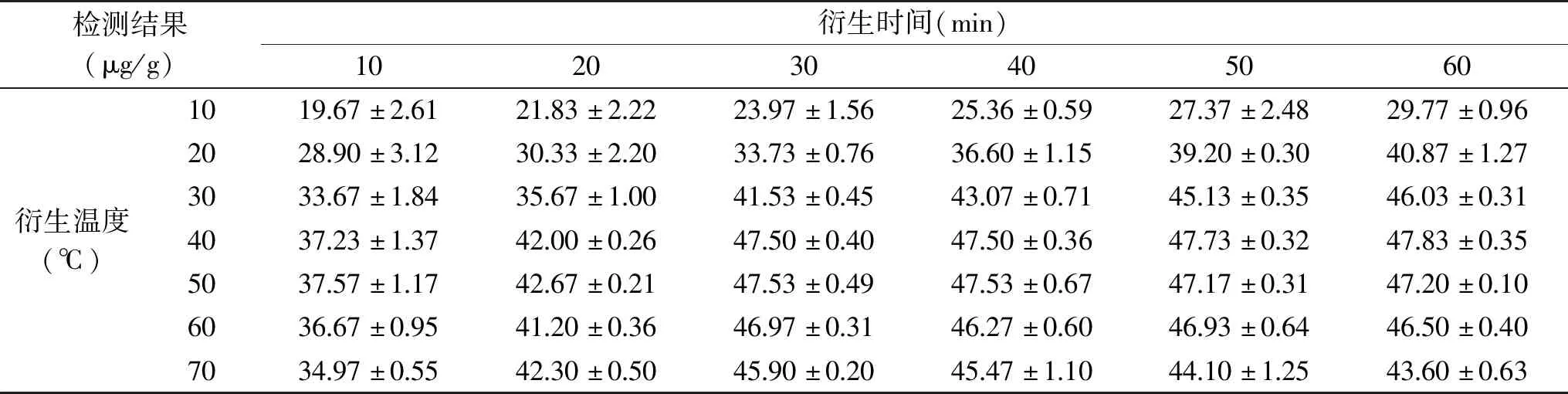
表2 不同衍生反应时间及衍生温度棋盘试验结果
由表2可知,当衍生温度为40 ℃,衍生时间为30 min时,测得样品中维生素C含量为47.5 μg/g,回收率为96.5%。完全满足检测要求。当反应温度超过40 ℃,反应时间大于30 min时,检测得到目标物含量并没有明显增加,反而由于时间过长,温度过高,导致衍生产物有微弱分解现象。所以选择最佳衍生温度为40 ℃,最佳衍生时间为30 min。

表3 线性范围,回归方程,相关系数,检出限
2.5 衍生液浓度优化
衍生试剂浓度对检测结果的影响,结果如图3所示。

图3 衍生液浓度对衍生反应的影响
由图3可知,随衍生液浓度的升高,衍生产物的含量逐渐增加,当浓度增加到1.0 g/L时,继续增加衍生液浓度,衍生产物含量并没有明显增加。因为当衍生液浓度在一定范围时,随衍生液浓度增加,衍生反应也会随之加快,一定时间内表现出的现象就是衍生产物含量增加。当衍生液浓度到达某一值时,衍生速度达到最快,继续增加衍生液浓度,并不能更好的加快反应速度,所以为节约成本,选用衍生速度达到峰值所对应衍生液的浓度1.0 g/L即可。
2.6 色谱条件选择
流动相pH环境选择,当流动相pH大于4时,色谱图中目标峰会变宽,保留时间变大(如图4A)。是由于色谱柱固定相载体球形硅胶对目标物的羟基有一定的吸附能力,当pH减小,酸性增强时硅胶吸附能力减弱,得到的色谱峰形状尖锐,对称(如图4B)。当流动相pH小于4时,保留时间变小,影响目标峰与杂质峰的分离效果(如图4C)。所以选择流动相pH环境为4.0。
流动相中有机相选择,当单独使用乙腈+水作为流动相时,该目标物色谱峰会产生前延现象(如图4D)。所以选用pH=4.0乙酸铵缓冲液(v)+乙腈(v)+甲醇(v)=80+15+5作为流动相时,避免前延和拖尾现象,保证峰形良好。

图4 不同流动相得到的不同色谱图
2.7 线性方程、线性范围、相关性系数、最低检出限
绘制维生素C标准溶液的工作曲线;该标准曲线的线性范围、回归方程、相关系数、仪器检出限(3倍信噪比)值见表3。
由表3可知,该方法线性范围宽,相关性系数良好,3倍信噪比(仪器检出限)低,样品处理方法为10倍稀释,所以该方法检出限为0.2 μg/g。较国标方法和已报道的文献方法更灵敏,完全适合痕量维生素C的检测。
2.8 回收率及精密度试验
按照1.2.3方法分别处理维生素C含量标识值为49.2、15.4、5.7 μg/g的阳性样品,每种样品平行处理8次,并检测其中维生素C的含量。计算每种浓度水平的平均回收率和相对标准偏差(relative standard deviation,RSD),结果见表4。
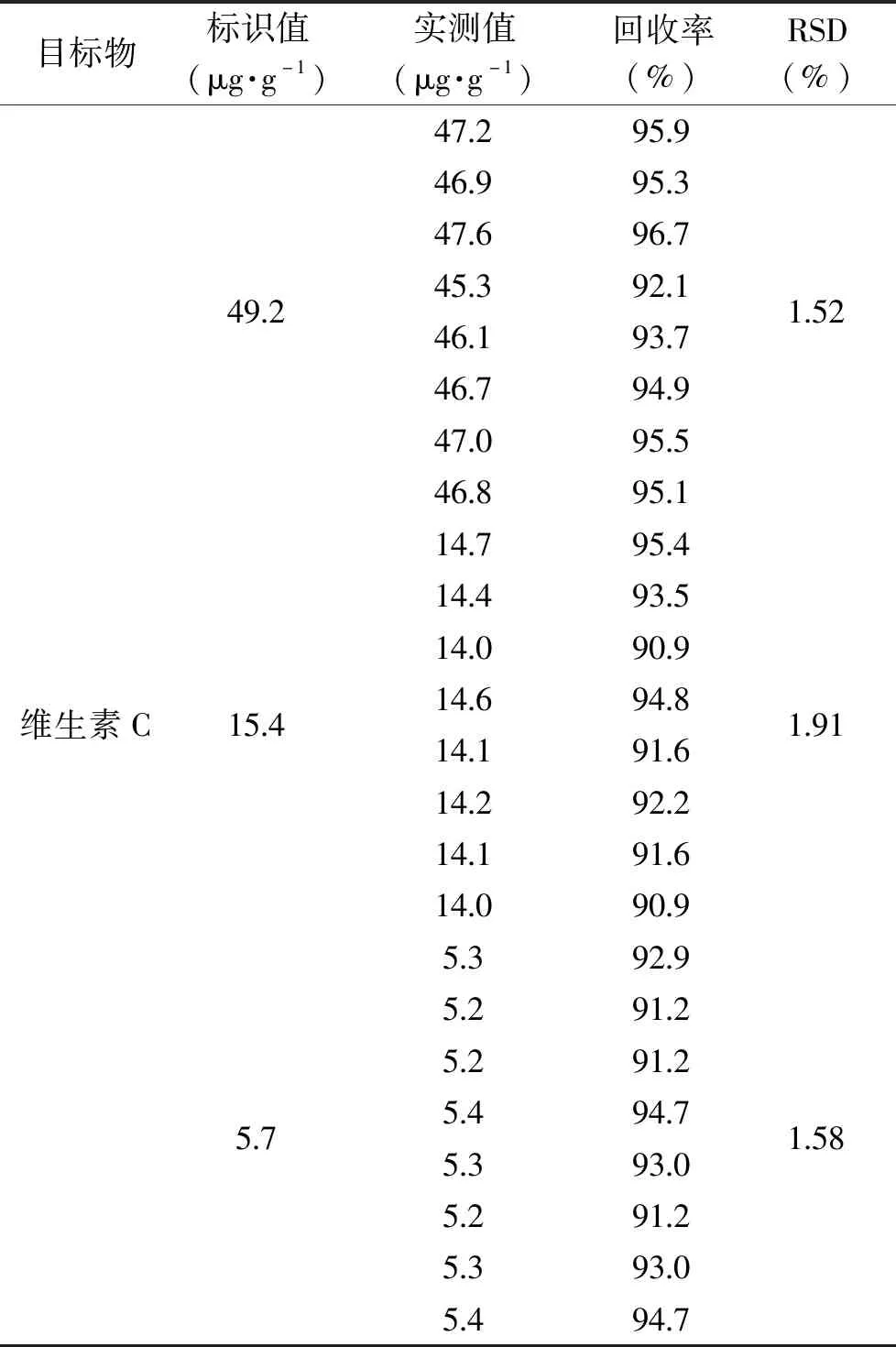
表4 3个水平下维生素C回收率试验结果
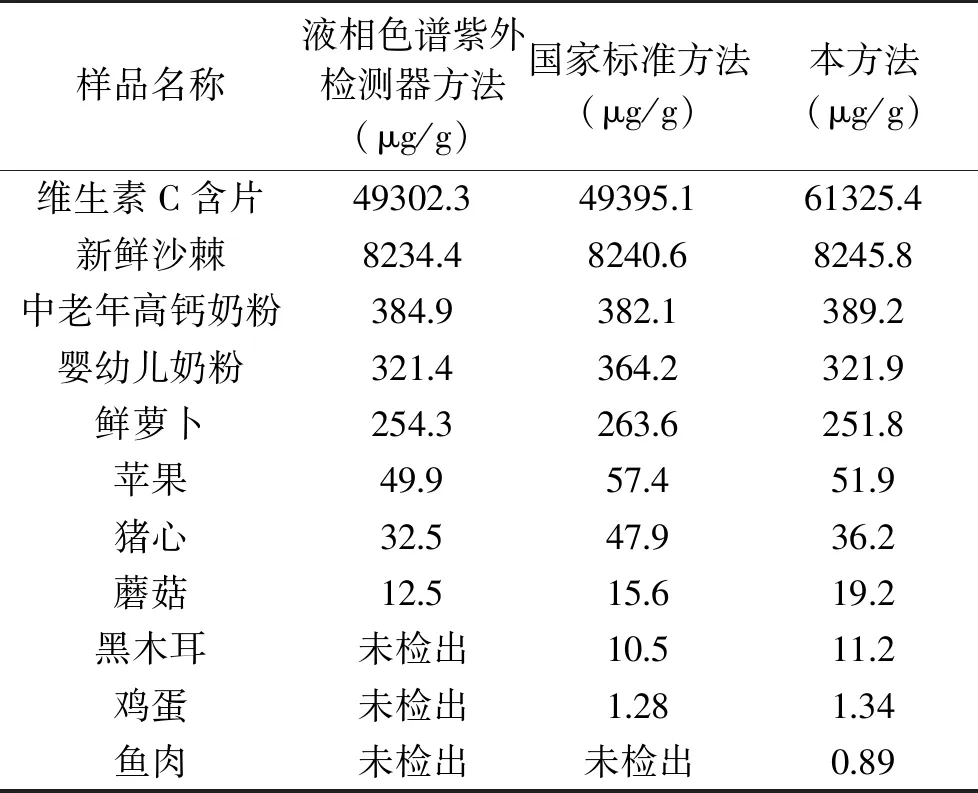
表5 不同方法对不同样品中维生素C含量测定结果
由表4可知,该方法回收率为90.9%~96.7%,RSD%≤2%,证明该方法结果稳定,满足检测要求。
2.9 实际样品测定及方法对比
分别采用本方法(方法检出限为0.2 μg/g),国标方法[6](方法检出限为1 μg/g)和液相色谱-紫外检测器方法[12](方法检出限为6 μg/g)对维生素C含量较多的样品(包括:乳粉、沙棘、新鲜果蔬等)和维生素C含量非常少的样品(包括:菌类、鱼类、鸡蛋等)进行测定,结果见表5。
由表5可知,液相色谱紫外检测器方法,收到检测器自身灵敏度限制,对含量10 μg/g以下样品无法准确测定。国标方法采用灵敏度较高荧光分光光度法,但受到衍生产物荧光强度限制,对含量1 μg/g以下样品无法准确测定;同时无法分辨所测得的荧光强度是由维生素C衍生产生的荧光,还是由样品中不易除去的杂质所产生的荧光,所以测量值不稳定;自建液相色谱荧光检测器方法,克服以上两种方法缺点,具有高灵敏度的同时,也降低背景及噪声对检测结果的影响,适合各类样品中维生素C的检测工作。
3 结论
结合液相色谱高自动化,高稳定性的优点。利用高灵敏度的荧光检测器,选择高强度荧光衍生试剂初步建立了一种实验操作简单,检测结果准确度高,稳定性良好,方法检出限低,灵敏度高,适合各类食品中痕量维生素C检测的方法。并将该方法实际应用在日常检测工作中,有望将该方法推广至其他领域,同时也为烯二醇类物质的荧光衍生方法及检测方法提供研究参考。
猜你喜欢
化工设计通讯(2022年10期)2022-12-31
波谱学杂志(2022年2期)2022-06-14
昆明医科大学学报(2021年8期)2021-08-13
昆明医科大学学报(2021年8期)2021-08-13
昆明医科大学学报(2021年3期)2021-07-22
农药科学与管理(2019年6期)2019-11-23
中央民族大学学报(自然科学版)(2018年3期)2018-11-09
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
现代检验医学杂志(2015年1期)2015-02-06
质量安全与检验检测(2012年6期)2012-09-17
