
分散固相萃取-超高效液相色谱-串联质谱法测定豆芽中6种喹诺酮类药物
2020-12-10 03:22:22张今君夏慧丽高海波
食品工业科技 2020年23期
张今君,夏慧丽,高海波
(台州市食品检验检测中心,浙江台州 318000)
豆芽是颇受百姓喜爱的芽菜类蔬菜之一,富含维生素C、矿物质、酚类化合物、游离氨基酸及微量元素,具有丰富的食用价值[1]。但豆芽在生产过程中极易滋生酵母、假单胞菌、大肠杆菌等微生物,易腐烂变质[2],因此一些不法商贩为了缩短豆芽生长周期、增强抗菌能力、改善外观品相,往往在豆芽生长过程中违规滥用植物生长激素、农药、抗生素等药剂,致使问题豆芽事件层出不穷。
目前,针对豆芽中赤霉素、6-苄基腺嘌呤、4-氯苯氧乙酸、2,4-2氯苯氧乙酸等植物生长调节剂的检测,我国已经发布了相关补充检验方法[3];针对多菌灵、涕灭威、咪鲜胺等农药残留的检测也有气相色谱-质谱联用法、液相色谱-串联质谱法等多个标准方法[4-6];针对喹诺酮类等抗生素药物的检测标准及文献多以动物源性食品为主[7-14],适用范围主要是畜禽肉、水产品及动物内脏等。而专门针对豆芽等植物源性食品中喹诺酮类等药物残留的文献相对较少,检测目标物相对单一,前处理方法主要是固相萃取、液液萃取等传统方法[15-21],成本较高,操作过程耗时、繁琐。因此,建立简便快速、经济有效,适于推广的豆芽中多种喹诺酮类药物残留的测定方法,对于解决当前豆芽行业滥用药物问题具有重要意义。
分散固相萃取法[22-24]简单、快速、廉价、高效、安全,是近年来发展较快的农产品检测样品前处理方法,本文应用分散固相萃取,结合兼具高灵敏度、高选择性的超高效液相色谱-质谱联用技术,建立豆芽中6种喹诺酮类药物残留检测方法。探讨了提取条件、净化填料用量、仪器条件参数、基质效应等影响因素,以期为豆芽中喹诺酮类等药物残留的检测标准建立提供依据。
1 材料与方法
1.1 材料与仪器
阴性豆芽样品 参照DB33 625.1-2007豆芽生产技术规程自培,绿豆(黄豆)经过清洗、杀菌(70~80 ℃热水烫豆1~2 min)和浸豆(20~25 ℃的清水,绿豆3 h,黄豆6 h)后进行培育(温度23~25 ℃,4 h淋水一次),4~6 d后采集豆芽样品;80批次测试样品 随机采集于菜市场、农贸市场和超市;恩诺沙星(Enrofloxacin,纯度99.0%)、环丙沙星(Ciprofloxacin,纯度94.8%)、氧氟沙星(Ofloxacin,纯度99.3%)、诺氟沙星(Norfloxacin,纯度97.2%)、洛美沙星(Lomefloxacin,纯度99.4%)、培氟沙星(Pefloxacin,纯度93.9%)、恩诺沙星-d5(Enrofloxacin-d5)、环丙沙星-d8(Ciprofloxacin-d8)、氧氟沙星-d3(Ofloxacin-d3)、培氟沙星-d5(Pefloxacin-d5)、洛美沙星-d5(Lomefloxacin-d5)、诺氟沙星-d5(Norfloxacin-d5) 纯度均大于95%,标准品,德国Dr.Ehrenstrorfer公司;乙腈、甲醇、甲酸 色谱纯,美国Merck;去离子水 Milli-Q系统纯净水;N-丙基乙二胺(PSA)、十八烷基键合硅胶(C18)、无水硫酸镁(MgSO4)、石墨化碳(GCB)分散固相萃取吸附剂 上海安谱实验科技股份有限公司;氯化钠 分析纯,天津市科密欧化学试剂有限公司。
QTRAP 5500三重四极杆-线性离子肼质谱系统 美国AB Sciex公司;UPLC液相色谱系统 日本岛津公司;Microfuge 20离心机、Allegra X-30R冷冻离心机 美国贝克曼库尔特公司;涡旋混匀器 德国IKA;2150 TH超声波仪 上海安谱;PR 1602 ZH/Z电子天平 奥豪斯电子天平有限公司。
1.2 实验方法
1.2.1 标准曲线绘制 标准储备液:准确称取每种标准品各10 mg(精确至0.1 mg),分别置于10 mL容量瓶中,用甲醇溶解并稀释至刻度,配制成浓度各为1 mg/mL标准储备液,-18 ℃保存。
混合标准中间液:准确吸取上述标准品及同位素内标标准储备液各25 μL置于25 mL容量瓶中,甲醇稀释至刻度,配制成浓度均为1 μg/mL的混合标准中间液,-18 ℃保存。
标准工作溶液:吸取混合标准中间液10、50、100、500、1000 μL于10 mL容量瓶中,加入同位素内标中间液50 μL,用50%乙腈水稀释至刻度得各目标物浓度为1、5、10、50、100 μg/L,内标物浓度为5 μg/L,现配。
按优化后条件进行测定,以标准品浓度为横坐标,标准品与内标物峰面积比值为纵坐标绘制标准工作曲线。
1.2.2 样品前处理 提取:称取粉碎均匀的豆芽样品10.00 g,置于50 mL具塞离心管中,添加200 μL同位素内标中间液,加入10 mL 1%甲酸乙腈溶液,涡旋振荡10 min,加入4 g氯化钠,涡旋混合1 min,在4 ℃下6000 r/min离心5 min,将上清液倾入50 mL具塞离心管,残渣再用10 mL1%甲酸-乙腈溶液重复提取1次,合并上清液于50 mL离心管。
净化:移取上层乙腈溶液1 mL置于装有PSA 50 mg、C18100 mg、GCB 10 mg、MgSO4150 mg的分散固相萃取净化管中,涡旋混合2 min,以6000 r/min离心5 min,取上清液0.5 mL加入0.5 mL水,涡旋混匀,经0.22 μm有机滤膜过滤后上机测试。
1.2.3 仪器条件 色谱条件:Waters ACQUITY UPLC BEH C18柱(100 mm×2.1 mm,1.7 μm),柱温:35 ℃,流速:300 μL/min,进样体积5 μL,流动相:A为含0.1%甲酸水溶液,B为甲醇。梯度洗脱条件见表1。

表1 梯度洗脱条件
质谱条件:电喷雾离子源(ESI源);正离子扫描(ESI+);多反应监测(MRM);电喷雾电压(IS)5500 V;雾化气压力(GS1)50 psi;辅助气压力(GS2)50 psi;气帘气压力(CUR)35 psi;离子源温度(TEM)550 ℃,各化合物定性离子、定量离子对及其碰撞能量等参数见表2。
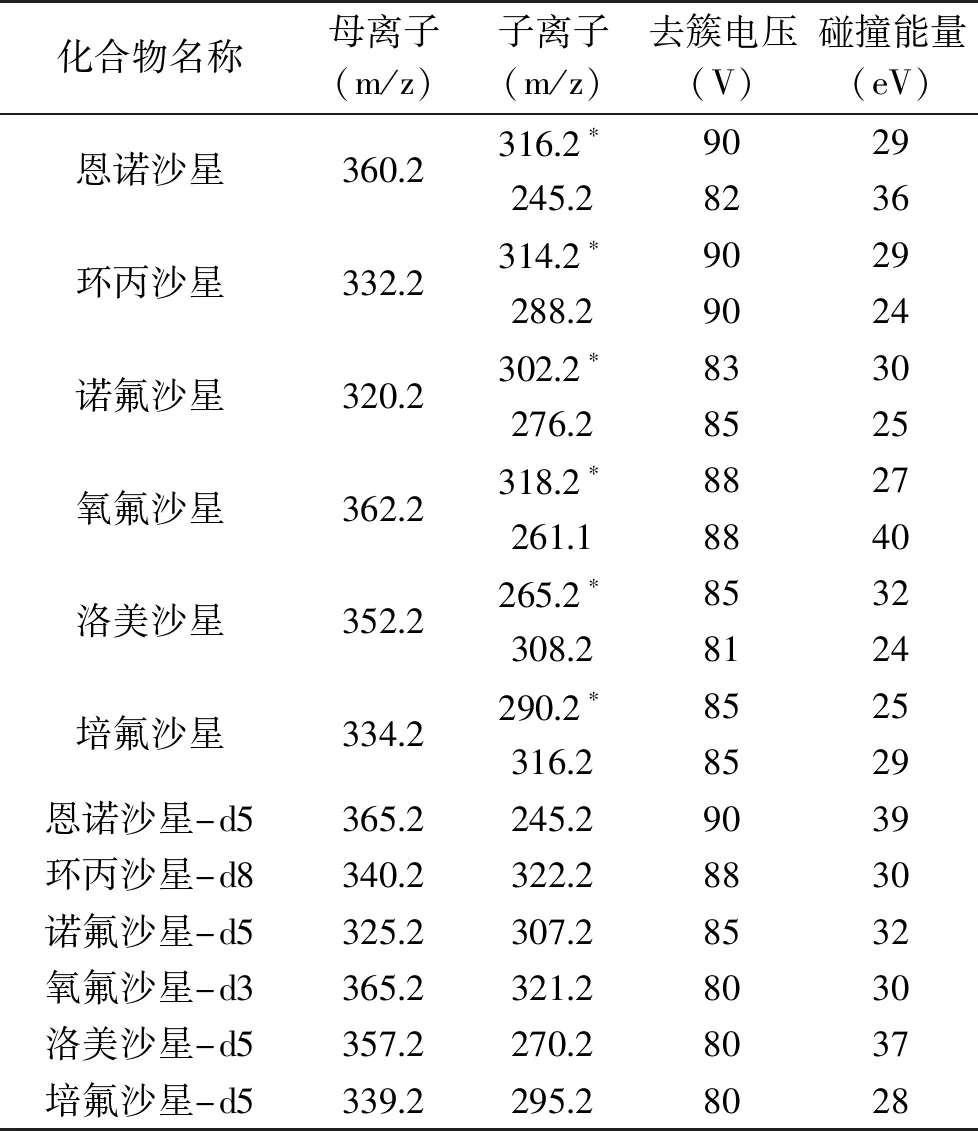
表2 化合物的质谱参数
1.3 数据处理
各组分通过计算机在Multi Quant 3.0.2软件上谱图处理,通过各组分的离子比进行定性确证,采取内标法计算各组分的相对含量。数据使用Micro-soft Excel 2007软件进行处理。
2 结果与分析
2.1 提取条件优化
2.1.1 提取溶剂的选择 喹诺酮类化合物分子结构中含有羧基和叔氨基,具有酸碱两性性质,易溶于酸性或碱性溶液。本实验采用乙腈、1%甲酸乙腈、1%乙酸乙腈、1%氨水乙腈各20 mL分两次对6种化合物进行提取,移取提取液0.5 mL,加入0.5 mL水涡旋混匀后过0.22 μm滤膜,检测回收率。添加浓度20 μg/kg,结果显示乙腈和氨水乙腈提取时各目标物回收率明显低于酸性乙腈,其中甲酸乙腈的回收率最高,均高于85%,结果见图1。实验选用1%甲酸乙腈作为提取溶剂。

图1 提取溶剂对6种喹诺酮药物回收率的影响
2.1.2 提取次数的选择 本文用20 mL1%甲酸乙腈分20 mL 1次,10 mL 2次,7 mL 3次三个方案对豆芽样品进行提取,移取提取液0.5 mL,加入0.5 mL水涡旋混匀后过0.22 μm滤膜,检测回收率。添加浓度20 μg/kg,结果显示,提取次数为2、3次时回收率均在89.90%~102.32%之间,各化合物提取效率高,结果见图2。考虑操作简便因素,选取10 mL 2次方案提取豆芽中化合物。

图2 提取次数对6种喹诺酮药物回收率的影响
2.2 净化条件优化
PSA、C18、GCB、MgSO4是分散固相萃取常用的4种填料。本文参照植物源性食品中农药及代谢物残留量检测的国家标准[25-26]设计不同用量对各目标物的回收率实验。在1.2.2其它条件不变情况下单独添加PSA、C18、GCB、MgSO44组填料各6个点进行回收率实验,添加浓度20 μg/kg,重复测定3次,填料用量及回收率见图3。PSA去除样品中的脂肪酸、糖等干扰物,对各目标物也有一定的吸附,在使用量50 mg之后对目标物吸附增加;C18去除样品中脂肪等非极性干扰物,在25~150 mg范围对各目标物吸附量稳定。GCB用量从10 mg上升至15 mg时各目标物回收率下降至60%以下;MgSO4在50~300 mg范围对各目标物吸附量影响较小。综合考虑回收率及除杂效果,选用PSA 50 mg、C18100 mg、GCB 10 mg、MgSO4150 mg组成分散固相萃取净化管,各目标物回收率较高。

图3 4种净化填料用量对6种喹诺酮药物回收率的影响
2.3 仪器条件优化
2.3.1 色谱条件的优化 喹诺酮类药物在正离子模式下进行测定,流动相中加入甲酸可以提高离子化效率,选择甲酸水溶液为水相流动相,对甲醇/甲酸水和乙腈/甲酸水进行比较,结果表明甲醇/甲酸水峰形尖锐,信号更高,本实验选择甲醇/0.1%甲酸水作为流动相,对梯度条件进行优化,采用优化后梯度洗脱方法,可以得到满意的实验结果。豆芽加标样品中提取离子色谱图见图4。
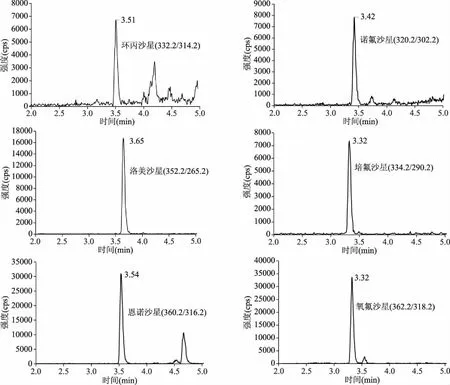
图4 20 μg/kg豆芽加标样品中MRM离子流图
2.3.2 质谱条件的优化 质谱条件优化时,去簇电压(DP)和碰撞能量(CE)这两个参数与化合物性质相关且影响较大。喹诺酮化合物中含有叔氨基,容易得到质子,因此选择正离子模式进行检测。实验将0.1 μg/mL各标准溶液经蠕动泵以7 μL/min流速进入ESI源进行质谱分析。通过Q1全扫模式获得分子离子峰,用子离子模式对分子离子峰进行分析,从打碎的离子中选取两个特征离子作为定性和定量离子。用MRM扫描模式优化DP和CE值,使得定量与定性离子强度响应最大,优化后的6种喹诺酮药物及同位素内标物质谱参数见表2。
2.4 基质效应评价
质谱分析中,由于流动相、目标物与样品中的其他成分同时进入质谱系统,进而影响了目标离子的离子化过程,使得目标物的质谱信号增强或者减弱,从而影响了分析结果的准确性和精密度[27]。本实验采用Guedes等[24]报道的方法对基质效应进行考察,在建立的前处理方法和仪器测定条件下,测定空白基质提取液与溶剂中同浓度目标物的响应值,通过目标物与内标物峰面积之比的相对比值评价。基质效应按照式(1)计算。6种喹诺酮药物在豆芽基质中的基质效应见表3,结果显示各化合物基质效应在0.97~1.04之间。本实验基质效应小,可选择溶剂配制标准工作溶液。
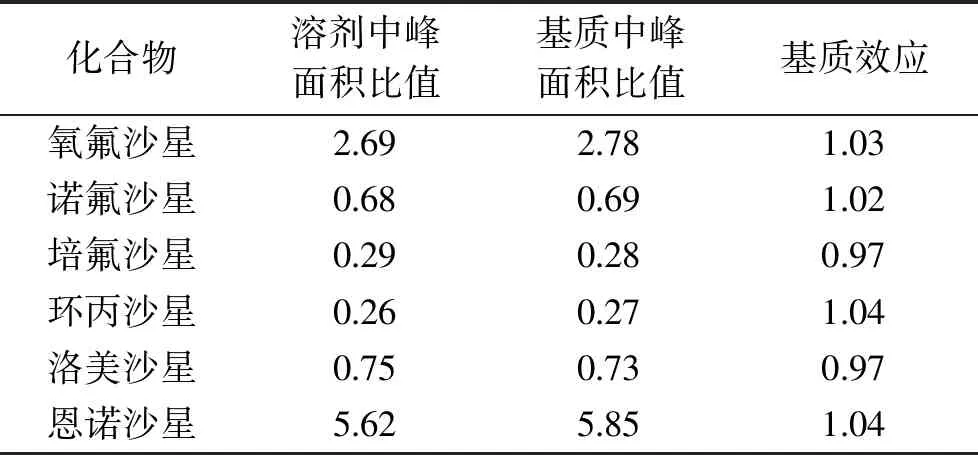
表3 6种喹诺酮药物的基质效应
式(1)
式中,ME表示基质效应的大小,当ME大于1.0时为基质增强,当ME小于1.0时为基质抑制;Am为基质提取液中化合物峰面积,Ami为基质提取液中同位素内标峰面积,As为溶剂中化合物峰面积,Asi为溶剂中同位素内标峰面积。
2.5 方法学验证
2.5.1 检出限、定量限和线性范围 将6种喹诺酮类药物的标准溶液稀释成不同的浓度,以标准品与内标物峰面积比值对应各自浓度进行线性回归,同时利用豆芽空白基质进行检出限和定量限的考察,以3倍和10倍信噪比确定6种喹诺酮类药物的检出限和定量限。结果表明,6种喹诺酮药物在1.0~100.0 μg/L的浓度范围内线性关系良好,相关系数均大于0.9982,检出限为0.1~0.3 μg/kg,定量限为0.3~1.0 μg/kg,具体见表4。

表4 6种喹诺酮药物的相关系数、检出限及定量限
2.5.2 回收率及精密度 在豆芽空白基质中添加不同浓度的6种喹诺酮类标准溶液进行加标回收实验,分别添加2、4、20 μg/kg三个浓度水平,每个样品平行测定6次,数据结果见表5。结果表明6种喹诺酮药物回收率均在96.53%~106.94%之间,相对标准偏差在2.73%~7.60%之间,符合GB/T 27404《实验室质量控制规范 食品理化检测》标准[28]要求。

表5 6种喹诺酮药物回收率及精密度(n=6)
2.5.3 实际样品测定 用建立的方法对从市场采集的80批次豆芽样品中的6种喹诺酮类药物进行测定,其中恩诺沙星、环丙沙星、诺氟沙星有检出,检出率分别为37.5%、26.2%、2.5%,氧氟沙星、培氟沙星、洛美沙星均未检出,具体情况见表6。

表6 80批次豆芽中6种喹诺酮药物检测结果
3 结论
本实验采用1%甲酸乙腈提取,PSA 50 mg、C18100 mg、GCB 10 mg、MgSO4150 mg分散固相萃取净化,同位素内标法定量,建立了同时测定豆芽中6种喹诺酮药物的高效液相色谱-串联质谱的分析检测方法。结果表明,各药物在1.0~100.0 μg/L浓度范围内的线性关系良好,相关系数均大于0.9982,三个添加水平的回收率范围为96.53%~106.94%,相对标准偏差为2.73%~7.60%,检出限为0.1~0.3 μg/kg,定量限为0.3~1.0 μg/kg。80批次豆芽样品中恩诺沙星、环丙沙星、诺氟沙星检出率分别为37.5%、26.2%、2.5%,氧氟沙星、培氟沙星、洛美沙星均未检出。本方法操作简单,准确度和精密度高,适合豆芽中喹诺酮类药物的定性和定量检测,可为芽菜类蔬菜中喹诺酮类药物残留的风险评估提供一种快速高效的分析手段。
猜你喜欢
铁军·少年国防(2023年8期)2023-10-03 14:57:25
世界最新医学信息文摘(2021年12期)2021-06-09 08:36:56
基层中医药(2020年7期)2020-09-11 06:38:02
中国蜂业(2018年4期)2018-05-09 06:25:08
国外医药(抗生素分册)(2016年5期)2016-07-12 14:25:37
国外医药(抗生素分册)(2016年2期)2016-07-12 14:25:01
当代化工研究(2016年6期)2016-03-20 16:21:46
中国卫生标准管理(2015年25期)2016-01-14 09:29:24
发明与创新(2015年29期)2015-02-27 10:39:44
小说月刊(2014年4期)2014-04-23 08:52:26
