
固态电解质与电极界面的稳定性
2020-12-05 07:34冯吴亮王飞周星吉晓韩福东王春生
物理学报 2020年22期
冯吴亮 王飞 周星 吉晓 韩福东 王春生‡
1) (复旦大学材料科学系, 上海 200433)
2) (复旦大学化学系, 上海 200433)
3) (Department of Chemical and Biomolecular Engineering, University of Maryland, College Park, MD 20742, USA)
4) (Department of Mechanical, Aerospace, and Nuclear Engineering, Rensselaer Polytechnic Institute, Troy, NY 12180, USA)
相比于有机体系锂离子电池, 全固态锂金属电池有望同时提高电池安全性和能量密度, 因而受到广泛的研究和关注. 固态电解质的电化学窗口决定了电解质在高压电池充放电过程中是否保持稳定. 目前的固态电解质, 热力学稳定电化学窗口较窄, 限制了其与高电压正极以及锂金属负极的匹配. 因而能否形成动力学稳定的界面, 决定了全固态电池是否能够持续高效工作. 本文总结归纳了固态电解质的热力学稳定窗口的实验和理论计算研究进展, 并对提高界面稳定性的实验进展进行了简述. 在此基础上, 提出构建动力学稳定性界面及防止锂枝晶的思路, 并展望了全固态电池界面构建的研究方向.
1 引 言
电解质在所有形式的电池中必不可少, 其基本作用是在正负极之间作为电荷传递的介质并防止电池短路. 目前的商用锂离子电池主要采用液态有机电解质, 存在易泄露、腐蚀性强、易燃易爆等缺点[1,2]. 随着现代科技的发展, 新型电子产品及新能源汽车等对锂离子电池能量密度提出了更高的要求. 不断提高的能量密度使得基于嵌入式正负极的锂离子电池能量密度逐渐接近其理论极限, 并也带来了日益严重的电池安全隐患[3-5].
全固态电解质相比于液态有机电解质在众多方面具有显著优势: 1)陶瓷基固态电解质不易燃,可以极大地减弱电池的安全隐患[6,7]; 2)固态电解质优异的机械性能有助于阻挡锂枝晶生长, 使金属锂负极的应用成为可能, 能够大幅提高电池的能量密度[8,9]; 3)全固态电解质没有流动、凝固和挥发的问题, 可以组装成电堆并在宽电池的工作温度区间工作[10]. 近年来以硫化物[11](Li10GeP2S12, LGPS)和氧化物[12](Li7La3Zr2O12, LLZO)为典型代表的固态电解质已经实现接近甚至超过商用锂离子电解液的离子电导率, 引发了近年来的全固态电池研究热潮. 尽管存在种种优势, 但是其在锂金属全电池中的表现依然差强人意.
在一个理想状态下的全电池中, 正极和负极的化学性质决定了电池的能量密度; 而位于正负极之间的电解质, 其离子传递的快慢则决定了电池的功率. 对于全固态电池来说, 在电解质体相离子电导率已经足够高的情况下, 电解质和正负极之间的界面及锂枝晶已然成为了全电池性能的限制因素. 全固态电解质/电极固-固界面的高阻抗是学界的关注热点, 因此大量研究工作集中于改善电解质与电极之间的接触[13,14].
然而, 将全固态电池分为正负极、电解质和界面的表述仍然过于简化. 早期的大量报道认为全固态电解质具有优于液态电解质的电化学窗口, 在电解质的热力学稳定性足够好的情况下, 电解质在电池运行期间不发生任何化学或者电化学变化, 此时所有法拉第过程都只在电极内发生, 只需要保证界面接触以便离子传递即可实现全电池性能的发挥.但是, 近年来的报道逐渐提出固态电解质存在电化学稳定窗口窄的问题[15], 在这一认知下, 便需要以动力学方式(界面钝化)而非热力学方式实现电解质的电化学稳定性, 来推动实现全电池的稳定工作. 而动力学稳定性会受到高电压正极的强氧化性和锂负极的强还原性的挑战, 因此如何构建稳定的电解质/电极界面, 是实现全固态电池稳定工作的关键. 本文总结归纳了关于固态电解质热力学和动力学稳定性的主要实验结果和理论计算进展, 并在此基础上梳理总结已发表的提高界面稳定性的办法, 提出对界面稳定性构建的思考, 为最终开发出高电压的全固态锂金属电池提供未来的发展方向.
2 固态电解质的热力学稳定性
电解质的热力学稳定性可以通过其发生氧化和还原反应之间的电压范围来量化, 即“电化学窗口”. 对于全电池而言, 理论上正负极材料的氧化还原电势都必须落入该电化学窗口内, 全电池才可以正常运行. 因此, 得到准确的电解质的电化学窗口, 是保证电解质材料及界面稳定的第一步.
2.1 固态电解质的电化学窗口
表1 所列为几种主流的固态电解质电化学窗口的报道值与理论计算值. 氧化物基固态电解质的电压窗口明显高于硫化物与卤化物基电解质, 由于其氧化电位主要取决于O2—离子的氧化, 所涉及的析氧反应在动力学上比较困难, 因此普遍具有较高的氧化电位. 然而NASICON 型电解质中的高价金属阳离子Ge4+, Ti4+等易被具有较强还原性的金属锂还原为低价阳离子甚至金属单质, 导致此类电解质还原电位较高. 硫化物电解质由于受限于S2—的氧化与阳离子(P5+, Ge4+等)的还原, 因此电化学窗口普遍较窄. 材料的氧化/还原电位计算通常基于材料发生氧化/还原反应时自由能的变化而非能隙或分子轨道理论中HOMO-LUMO 能量之差. 大规模计算发现, 固态材料电化学稳定性主要受以下三个因素的影响: 1)材料中的阴离子对氧化和还原电位具有主导性的影响, 总体趋势为氮化物 < 氧化物 < 硫化物 < 氟化物; 2)高价阳离子对还原电位具有趋势性的影响; 3)根据材料中的锂浓度的不同, 富锂材料通常具有较低的氧化和还原电位, 贫锂材料则通常具有较高的氧化和还原电位[28].
在过去较长一段时间内, 无机固体电解质被认为具有较宽的电化学窗口, 以LGPS 与LLZO 为典型代表, 其电压窗口分别被认为达到0—5 V 与0—6 V, 然而理论计算表明其各自的实际电压窗口远低于过去的认知. 例如, 第一性原理计算发现LGPS 在0—1.7 V 与2—2.5 V 具有可逆的还原与氧化反应, 证明了其真实的热力学稳定窗口应远小于0—5 V[29]. 而LLZO 在2.91 V 时便会形成Li2O2,Li6Zr2O7与La2O3, O2则会在在高于3.3 V 时从Li2O2分解出, 在低于0.05 V 时被还原为Li2O,Zr3O, La2O3, 甚至形成Zr0(<0.004 V)[17]. 过去人们对固体电解质界面问题的关注点主要集中于界面接触的改善、电极与电解质之间的化学稳定性以及消除空间电荷层的影响, 而对固体电解质真实电压窗口缺乏深入研究, 导致电解质常在高于各自的电压窗口的范围下运行, 从而引起一系列界面副反应, 尤其是匹配了高压正极后其反应产物多为离子不良导体, 锂离子传输受到阻碍[30]. 除此之外, 其可逆的氧化还原反应亦会引起体积应变, 导致界面接触恶化, 界面阻抗进一步增加, 故加速了电池的电化学性能衰减. 因此, 固态电解质的电化学窗口到底如何? 究竟是像很多文献报道的宽化学窗口,还是如热力学计算所说的很窄的窗口? 如何解释理论和实验观测的分歧, 以及如何正确测量电化学窗口需要进一步深入探究.

表1 各类固体电解质电化学窗口的理论计算值与报道值概括Table 1. Summary of the theoretical calculations and the reported values of electrochemical windows for different solidstate electrolytes.
如图1 所示, 固态电解质的电化学窗口通常采用锂作为参比电极和对电极, 以铂或金作为工作电极, 采用循环扫描伏安法(CV)测试获得. 由于任何电化学反应的法拉第电流都与电极的面积、氧化还原活性物质的浓度及扩散系数和转移的电子数成正比, 因此采用平板电极作为半阻塞电极的电池结构所造成的电解质与集流体之间的接触面积小,会导致电解质分解的动力学缓慢, 因而导致反应电流过小, 无法被直接观测到. 如果反应生成的界面产物产生钝化效应, 会进一步减少电流并引起严重的极化, 导致实测值中的还原电位偏低与氧化电位偏高. 如图1 中的界面放大示意图所示, 固态电解质由于表面不是绝对意义上的平整, 与电极具有较大的间隙, 电极仅能从有限的点接触位置给予/接收电子. 而在理想情况下的, 界面接触应为完全的面接触以保证充分的氧化还原反应.

图1 (a) 传统固体电解质的循环扫描伏安法测试装置示意图; (b) LGPS[22], (c) LLZO[24]以及(d) Li2OHCl[31]电解质传统CV 测试曲线Fig. 1. (a) Schematic diagram of conventional cyclic scanning voltammetry device for solid-state electrolyte; CV testing curves for(b) LGPS[22], (c) LLZO[24] and Li2OHCl[31] solid-state electrolyte.
除此之外, 由于阴极与阳极界面接触状态的差异, 以及各自氧化还原产物的离子/电子电导率的差异, 造成了电解质的氧化还原电流远小于锂沉积-剥离电流大小. 平板状半阻塞电极与电解质有限的点接触导致氧化电流通常仅为微安级别, 而当电位负向扫描至零伏以下时, 在阳极处发生锂沉积行为的过程中界面接触有愈加牢固的趋势, 锂沉积电流甚至可达毫安级别, 导致原本微弱的电解质的氧化还原电流更加难以辨别. 如图1(b)—图1(d)所示, 由于电解质的氧化还原电流相比锂沉积电流极其微弱, 在整个扫描区间无法被识别出, 因此造成了初期研究中人们对LGPS, LLZO 以及反钙钛矿型(Li2OHCl)电解质电化学窗口, 尤其是对氧化电位的误判. 因此, 为了提高测量的准确性, 测量装置的改进点应主要集中于电极/电解质接触面积的增加, 尤其是电解质/半阻塞电极界面的改进,并保持在大于0 V 范围内进行分段扫描, 避免较大的锂沉积电流的干扰.
2.2 真实电化学窗口的测试方法
由于电解质/电极的接触面积是导致电解质电压窗口实测值远大于计算值的主导因素, 针对传统固体电解质电化学窗口测试所面临的问题, 在我们的早期工作中提出, 使用固态电解质和导电碳混合而成的复合电极, 将电解质/电极的接触由传统的平板电极间的二维接触实现了三维接触, 可以提高固-固界面的接触面积和反应动力学, 从而测得固态电解质真实的电化学窗口. 此方法测得的窗口与热力学计算结果相符合[25]. 相比于半阻塞电极, 这种混合电极能更真实地代表和反映全固态电池中的界面状况.
图2 所示为更精确地测试电解质电化学窗口的电池结构示意图, 通过将导电碳与电解质混合,极大地增加了电解质与电极的接触面积并加速了电解质分解的动力学过程, 电解质氧化后电子被更加有效地收集, 氧化电流获得明显提高, 同时牢固的面接触极大地降低了界面电荷转移电阻, 极化作用受到了有效抑制. 首先通过对采用了传统半阻塞电极的Li/LGPS/Pt 进行分段扫描(0—2.5 V 与2.5—4.0 V)发现了微弱的还原/氧化电流, 进而将LGPS 与导电碳按3∶1 质量比混合, 复合电极中电解质与电子导体的充分接触可有效避免动力学势垒阻碍LGPS 分解反应的发生, 从而更好地探究了LGPS 本身的热力学稳定性. 通过对Li/LGPS/LGPS-C/Pt 进行分段扫描(0—2.0 V 与1.0—3.5 V),分别于1.6 与2.1 V 发现了还原/氧化峰. 又通过铂黑代替碳组装了Li/LGPS/LGPS-Pt/Pt 电池以排除可能存在的C 与LGPS 发生化学反应的干扰,在同样的扫描区间进行了重复验证, 得到了相同的结果, 因此进一步证实LGPS 的电化学稳定窗口远窄于0—5 V. 类似的方法也被Swamy 等[32]采用, 并获得了类似的结果. 由于没有钝化层产生并且氧化和还原反应的可逆性高, 我们进一步发展了一种基于LGPS 电解质本身氧化还原反应的单一材料的全固态电池[23], 正负极均采用LGPS 与导电剂混合以获得优异的离子/电子电导率以供电池运行. 通过对比Au/LGPS/Au 在不同电位下的阻抗谱发现, 其奈奎斯特图由新制备时所表现出的典型的电解质阻抗谱经充电至2.7 V 变为了典型的电池阻抗谱, 从另一个角度验证了LGPS 电解质较窄的电化学窗口. 相应地, 我们采用了相同结构的Li/LLZO/LLZO-C/Pt 电池研究了LLZO 的真实电压窗口, 由于LLZO 还原电位接近0 V, 因此还原电流易受锂沉积电流的影响难以分辨, 而在4.0 V 发现了可辨识的氧化峰, 证实了LLZO 的真实氧化电位远低于过去的认知(6.0 V). 然而相比理论计算值, 测量值依旧存在大于1 V 的过电位,尽管LLZO 与碳的质量比调整到了2∶3, 由于LLZO 弹性模量较高, 与碳混合后仅靠加压方法难以达到硫系电解质-碳混合电极的接触状态, 同时,LLZO 的氧化过程涉及到动力学较差的固相到气相(氧气)的转变, 因而造成了较大的过电位.
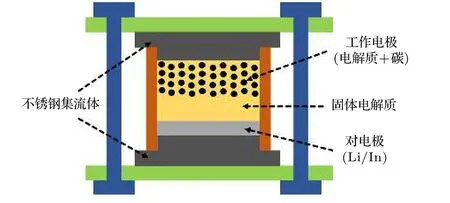
图2 电解质电化学窗口测试装置结构示意图Fig. 2. Schematic diagram of the testing device for the electrochemical stability window.
3 固态电解质的动力学稳定性
3.1 界面稳定性的重要性
相比于针对固体电解质离子电导率的提升, 现今固态电池的研究热点更集中于电解质/电极界面这一短板. 除了界面刚性固-固接触引起的巨大阻抗外, 对界面稳定性的研究更多的还是从抑制空间电荷层与电解质/电极化学反应副产物的角度入手, 然而对于电解质因电压窗口被严重高估而造成的界面不稳定现象的研究依旧未引起高度重视. 热力学计算表明, 大多数固态电解质不具备对锂金属或高电压正极材料的热力学稳定性. 三个机制可以导致界面的反应[33]: 1)电解质本身的电化学稳定性; 2)电解质与电极的化学反应; 3)电解质和电极在特定电压下的电化学反应. 界面处的反应会导致界面层的生成, 而不同的界面层性质则决定了不同的界面兼容性. 当电极/电解质的化学势不匹配时,即热力学不稳定时, 两种材料之间一旦接触则会自发发生化学反应. 由于动力学稳定性主要关注热力学不稳定的过程中反应的速率问题. 因此本文中提到的界面动力学稳定则是基于热力学不稳定的前提下, 反应速率较慢的情形. 若反应产物为离子导体而电子绝缘, 则只是以增大的电荷转移电阻为代价换取动力学稳定, 从而拓宽了电解质的工作电压范围. 然而, 若产物为离子电子混合导体, 则两者之间的化学反应会持续不断地进行. 理想情况下,界面层具有钝化效应, 可以延迟甚至阻绝反应的进一步发生, 从而保证了固态电池稳定的循环性能.这一机制与商用锂电池中的钝化膜(SEI)的效应类似[34].
按电解质/电极界面的化学稳定性可分为如图3 所示的三大类:
1)第一类为热力学稳定的界面, 即两者化学势相同, 不存在自发反应, 无中间层生成的理想界面. 符合此类界面的电解质材料较少, 尤其是对锂金属稳定的材料大多不具备高离子电导率, 或不满足高氧化电位的要求;
2)第二类为热力学与动力学均不稳定界面.此类界面生成的副产物中间层为混合导体界面, 即同时具有电子和离子导电性, 从而导致电解质体相通过此中间层不断地得失电子, 副产物中间层不断地向电解质一侧生长侵蚀原电解质体相, 在负极亦可能导致锂枝晶的产生造成严重的自放电甚至短路, 故此类界面应当避免. LGPS 或Li3MX6(M=In, Y, Er,X= 卤素)型卤化物电解质与金属锂接触后, 锂金属能够将电解质中的高价阳离子Ge4+,In3+, Y3+, Er3+等还原, 还原产物除Li3P, Li2S, LiX外还包括Li-Ge, Li-In(待验证)等合金. 由于合金相的存在, 其混合导电的性质无法对界面实现钝化保护, 于是分解反应会持续发生. 类似的机制也发生于NaSICON 型电解质(Li1+xAlxTi2x(PO4)3, LATP;Li1+xAlxGe2x(PO4)3, LAGP)与金属锂的界面. 而硫化物固态电解质与氧化物正极材料之间亦存在不同的化学势, 反应产物往往包含电子导电的过渡金属硫化物(如硫化钴), 同负极界面所面临的问题一样, 亦无法提供钝化保护, 造成不兼容性. 热力学分析发现, 界面反应的主要驱动力是两者之间阴离子的化学不兼容所导致的. 尽管氧化物固态电解质与氧化物正极材料的化学兼容性相对较好, 但为了改善其较差的物理接触, 高温处理后依然会生成显著的界面层.

图3 三类电解质/电极界面化学稳定性示意图Fig. 3. Schematic diagram of chemical stability of the three kinds of electrolyte/electrode interfaces.
3)第三类为热力学不稳定而动力学稳定界面.尽管大多数固态电解质对锂金属不具有热力学稳定性, 但具有锂离子电导率且电子绝缘的钝化界面层形成后依旧能够实现界面的稳定, 类似于常见的SEI 膜或CEI(cathode electrolyte interphase)膜等. LiPON 薄膜虽然本身对锂金属不具有热力学稳定性, 其被锂还原的反应产物主要为电子绝缘的Li3N, Li3P, Li2O 等, 这一反应形成的界面层不仅对锂金属稳定, 而且具有钝化作用. 此外, Li3Ns等本身亦是快离子导体材料, 其高离子电导率有利于界面层的锂离子输运, 从而实现低界面电阻.SEI 膜的物质含有LiF, Li3N, Li2O 等常作为动力学稳定界面. LLZO 由于表面有稳定的Li2CO3钝化层, 可一定程度上提高氧化电位, 在负极一侧的还原反应由于其热力学驱动力只有0.05 eV, 在实际过程中很有可能会被动力学因素所抑制, 因此也可归类为第三类界面层. 而硫化物电解质与硫电极的化学兼容性相对优越, 因此基于硫化物电解质的锂硫电池有很大发展潜力.
电流密度下可运行超过500 h, 界面稳定性大大提高. Zhao 等[38]将氟引入到以硫银锗矿型Li6PS5Cl(LPSCl)电解质中, 制备了氟化的硫化物电解质LPSCl1—xFx(图4(b)), 锂金属兼容性得到了显著提升的主要原因得益于原位形成致密的LiF 界面层.
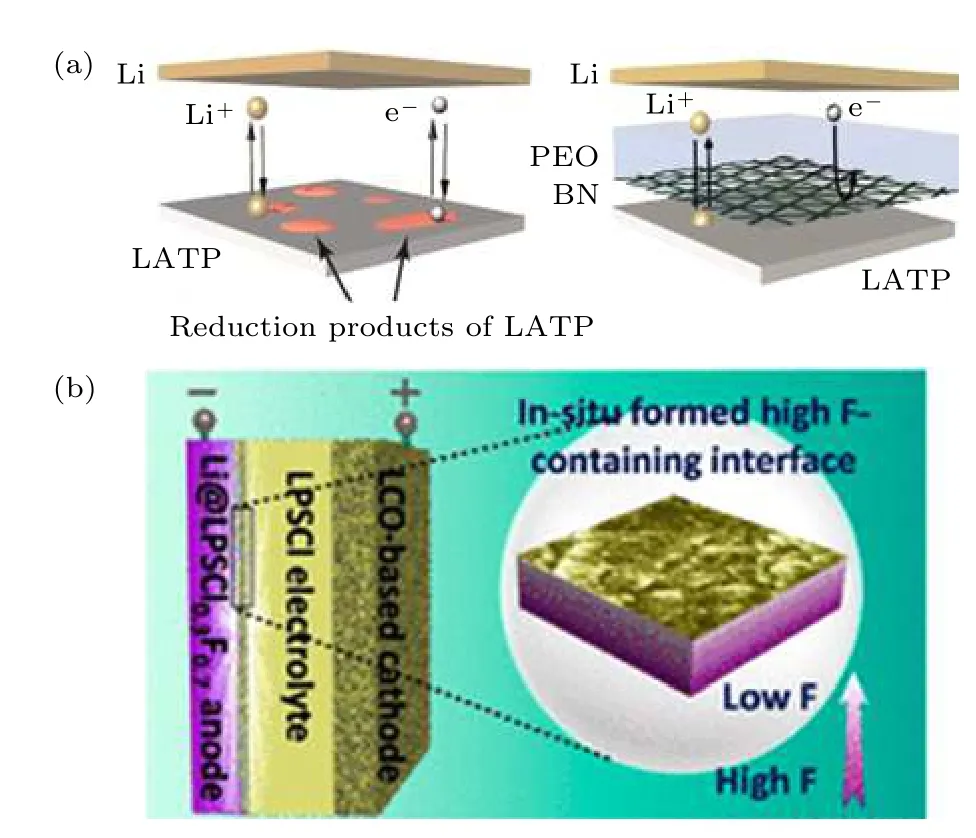
图4 (a) 氮化硼(BN)中间层稳定LATP/Li[37]与(b) LiF中间层稳定LPSCl/Li[38]界面示意图Fig. 4. Schematic diagram of the (a) LATP/Li interface stabilization by BN[37] and (b) LPSCl/Li interface stabilization by LiF[38], respectively.
3.2 界面的(电)化学稳定性
3.2.1 负极界面稳定性
由于金属锂具有很高的锂原子电化学势, 除了常见的以LiF, Li3N, Li2O 为代表的SEI 膜主要成分外, 几乎所有固体电解质对锂均热力学不稳定.其中LLZO 具有较低的对锂还原电位, 常被认为是对锂稳定材料. 然而Zhu 等[35]通过XPS 测试与理论计算发现, LLZO 与金属锂常温下接触后即发生了Zr4+→Zr2+→Zr0的还原过程, 并对比了Al3+,Ta5+, Nb5+三种掺杂元素的还原行为, 发现Nb5+的还原电位相对最高, Nb5+掺杂的LLZO 出现了明显的“氧缺失层”(oxygen-defcient interphase, ODI),但由于Zr0与Al3+, Ta5+, Nb5+的还原态产物极其微量, 因此对电解质性质的影响较小, 仅有Nb5+掺杂的LLZO 在与金属锂接触后, Li/LLZO/Li 对称电池阻抗随时间推移出现略微的增加.
NASICON 型、钙钛矿型、以及LGPS 电解质由于高价阳离子易被金属锂还原为单质后形成合金, 生成上述的第二类界面, 常采用引入具备第三类界面特征的缓冲层或合金负极解决与金属锂的接触问题. Yan 等[36]发现除常见锂卤化物外, BN具有优异的对锂稳定性与合适的机械强度, 将其作为人工引入的SEI 膜修饰金属锂可获得优异的电化学性能. Cheng 等[37]通过化学气相沉积法在锂负极上沉积小于10 nm 的氮化硼薄膜作为界面保护以隔绝LATP 与金属锂(图4(a)), 并分析对比了LATP 的失效机理和BN 的保护作用. 通过Li/Li 对称电池发现裸露LATP 在81 h 后即发生短路,而在有BN 保护的情况下, 对称电池在0.3 mA·cm—2
3.2.2 锂枝晶的生长与抑制
固体电解质与锂金属界面反应被认为与锂枝晶的形成和生长有重要关联. 固体电解质与锂金属发生反应, 既降低锂沉积/剥离的库仑效率, 生成不均匀的界面相以及裂纹, 也会导致不均匀的锂成核, 从而为锂枝晶的生长提供了先决条件. 如果生成界面是有电子导电性的, 锂枝晶甚至可以直接在电解质内部晶界或空隙中沉积生长. 氧化物电解质由于易生成Li2CO3疏锂层, 其与金属锂界面较高的表面能导致熔融锂的润湿性较差, 因而造成了较大的界面阻抗. 为减小表面能以增加金属锂在电解质表面的浸润性, 近年来大量关于亲锂层修饰界面以增加熔融锂在电解质上的浸润性, 以及与电解质亲和性较强的负极材料被广泛报道, 包括Au[39],Ag[40], Si[41], Ge[42], Sn[43], Al2O3[44], ZnO[45]等修饰层, 以及Li-Al[46], Li-C[47]等与LLZO 表面亲和性较强的负极材料. 然而由于表面能的降低, 此类材料在增强了负极浸润性的同时, 亦增加了锂枝晶垂直生长的风险. 我们课题组的研究表明, 通过调控界面相与锂金属的界面能, 如形成界面能较大的疏锂LiF 界面, 可以抑制锂沉积的垂直生长, 从而有效抑制锂枝晶的生成[48-52]. 但是疏锂界面又带来与金属锂的接触问题. 通过金属锂与电解质反应生成界面层, 原位生成紧密接触的疏锂界面, 将是解决这一矛盾最有效的办法, 比如直接在锂与电解质生成疏锂离子导电相Li3N[53], LiF[54], 既可降低界面阻抗也可抑止锂枝晶. 此外, 为消除疏锂层的存在而导致的界面接触不牢固问题, 也可以采用金属锂与修饰层的转化反应以获得一个维持界面牢固接触的中间层, 如近年来报道的基于转化反应的Cu3N[55], SnF2[56], MoS2[57]等修饰层, 均获得了界面阻抗的显著降低与临界电流密度的提升. 其中,在Cu3N 与金属锂的转化反应中, 作者指出其铜纳米颗粒产物可均匀地分布在石榴石电解质与金属锂的界面上. 铜颗粒的均匀分布增强了锂金属负极表面电势的均匀性, 使得锂沉积更加均匀, 因而提高了锂对称电池的电流密度, 实现了对锂枝晶的抑制作用.
3.2.3 正极界面稳定性
在正极界面一侧, Banerjee 等[58]将活性物质LiNi0.85Co0.1Al0.05O2(NCA)与硫化物电解质Li6PS5Cl 物理混合并充电到4.3 V, 经XRD 表征发现, 即使是原始的NCA 与电解质混合也会产生Li3PO4与Ni3S4, 而充电到4.3 V 的NCA 更是与Li6PS5Cl发生严重的反应, 生成Li3PO4, Ni3S4与LiCl. 而Ni3S4具有电子导电性, 会使两者之间的界面反应持续进行. Zhang 等[59]也使用XPS 技术在循环后含有LiCoO2, Li10GeP2S12与C65 的复合正极中检测到了SOx与GeO2. 此外, Auvergniot 等[60]使用XPS 与扫描俄歇电子显微镜(SAM)表征了Li6PS5Cl 与LiCoO2, LiNi1/3Co1/3Mn1/3O2, LiMn2O4三种活性物质之间的界面反应, 结果表明, Li6PS5Cl对这三种活性物质的反应性按照以下顺序增加:LiCoO2< LiNi1/3Co1/3Mn1/3O2≪LiMn2O4. 为了增加LLZO 电解质与LiCoO2接触, Park 等[61]将LLZO 与LiCoO2于700 ℃下共烧结后发现Li7La3Zr2O12从立方相到四方相的转变与La2CoO4锂离子绝缘扩散相的生成. Vardar 等[30]发现此扩散相在300—500 ℃温度下即可生成, 从而导致电化学性能恶化.
硫化物电解质的氧化电位较低, 其不可逆氧化过程在阻碍离子的传输、增大电荷转移阻抗的同时也导致了全固态电池的首圈库仑效率低, 进而影响了电池的循环寿命[17,25]. Koerver 等[62]使用XPS表征NCM811 与硫化物电解质界面时发现, 随着充电截止电压的升高, S 2p 信号中的峰向高结合能处位移, 表明电解质的电化学分解随着电压的升高而增加, 这也伴随着Nyquist 图中归因于正极/电解质界面的中频半圆逐渐增大. 而随着刻蚀深度的增加, 分解产物逐渐减少, 表明靠近集流体处的电解质分解更加严重. Dewald 等[63]使用电位范围逐渐增加的逐步循环伏安法测定Li10GeP2S12的电化学窗口, 以铟作为参比电极和对电极, 如图5所示. 当施加较低的反转电位时电解质是稳定的,当反转电位逐渐增高时出现了归因于电解质氧化分解的氧化峰, 而在之后的扫描中, 原本稳定的电位范围内出现了新的氧化还原峰, 这表明电解质的氧化分解产物发生了氧化还原反应. 而新出现的氧化还原峰可以归为Sx与之间的氧化还原反应.而从这种方法得到的Li10GeP2S12氧化电势(约2.6 V)要大于理论预测得到的氧化电势(2.14 V)[17],这说明电解质的电化学窗口不止由热力学来决定.Schwietert 等[15]通过XRD 观察到Li6PS5Cl 的脱锂相(Li4PS5Cl), 证明电解质是通过电解质的锂化与去锂化再分解成热力学稳定的产物, 而不是直接分解成热力学稳定的产物.

图5 分别使用铟和固体电解质/C 复合材料作为对电极和工作电极, 对Li10GeP2S12 进行循环伏安(0.1 mV·s—1)测试图[62]Fig. 5. CV curve (0.1 mV·s—1) of Li10GeP2S12 with Indium counter electrode and solid electrolyte/C composites working electrode[62].
由于电解质与正极之间Li+的化学势不同,Li+会自发从化学势高处向化学势低处运动[64], 从而导致界面处Li+浓度降低甚至形成一个锂耗尽层, 结果是界面处缺乏足够的可移动Li+导致界面处的高极化电阻[65,66]. de Klerk 等[67]建立了描述空间电荷层的模型, 表明了LiCoO2与Li7La3Zr2O12和LiCoO2与Li1.2Al0.2Ti1.8(PO4)3之间的空间电荷效应导致了纳米级的空间电荷层, 其贡献了小于1 Ω·cm2的界面阻抗. 但是作者同时也强调, 空间电荷层所导致的影响在不同材料之间可能会有很大的差异, 例如形成厚的或Li+完全耗尽的空间电荷层, 会显著地增强界面阻抗. Zhang 等[68]则通过6Li 二维核磁交换谱(6Li 2D-EXSY)验证了空间电荷层对LixV2O5与Li1.5Al0.5Ge1.5(PO3)4界面处Li+跨界面交换的影响, 当无空间电荷层时, Li+跨界面交换的活化能为0.315 eV, 当存在空间电荷层时这个值会迅速提升到0.515 eV, 从而额外增加了电荷转移电阻.
4 界面的力学稳定性
电解质/电极的界面力学稳定性取决于界面接触的稳定性, 由于电极材料在充放电过程中难以避免地发生体积应变, 界面应力的改变有可能导致界面接触面积降低, 使得电荷转移电阻增加, 降低电池的循环性能. 常见电解质与电极材料的杨氏模量如表2 所列, 硫化物电解质与反钙钛矿电解质由于具有较低的杨氏模量, 一方面采用简单的常温下加压方式即可实现电解质/电极牢固的面接触, 另一方面其优异的延展性可对电极材料充放电过程中产生的局部畸变进行缓冲, 因而具有较好的界面力学稳定能. Li3MX6(M= In, Y, Er,X= 卤素)型卤化物电解质由于具有较软的质地, 因此同样可采用冷压法制备全固态电池[74-76]. 由于氧化物型电解质与氧化物正极材料同时具有较高的杨氏模量,因此无法通过常温下的加压方式解决其固-固刚性点接触问题[12]. 为了增加电解质/电极接触面积,Hansel 等[77]以淀粉为模板剂构筑了3D 结构的LLZO 表面, 增加了LLZO 与Li4Ti5O12以及LiMn1.5Ni0.5O4的接触面积. Yan 等[78]采用流延法制备了LLZO 与LiFePO4薄膜, 通过共烧结后获得了紧密的接触. 小粒径材料具有较高的表面能, 尽管通过高温烧结的方式软化接触界面可一定程度上增加界面接触面积, 提高正极材料致密度, 但研究表明[30,79], 在低于500 ℃的温度下过渡金属氧化物正极中的过渡金属元素会与LLZO 发生相扩散,生成锂离子绝缘相LaMO3(M= Ni, Co, Mn)与失锂相La2Zr2O7, 并诱发立方相向四方相的转变.NASICON 型电解质采用相同的处理方法亦会导致LiMPO4(M= Ni, Co, Mn)扩散相以及分解相Li3PO4与AlPO4的生成, 增加界面阻抗[80]. Ohta等[81]采用硼酸锂作为助烧结剂实现了LLZO 与LiCoO2良好的面接触, 硼酸锂具有较低的熔点(700 ℃), 可降低共烧结的温度, 同时具有一定的离子电导率(2 × 10—6S·cm—1)以保证锂离子的传输, 此外, 助烧结剂的引入可有效隔绝LLZO 与LiCoO2, 抑制了烧结过程中扩散相的生成. Han 等[82]将此方法更进一步, 引入的Li2.3C0.7B0.3O3可与LLZO 和LiCoO2表面自然生成的Li2CO3钝化层原位反应, 生成同样具有一定离子电导率与较低熔点的Li2.3—xC0.7+xB0.3—xO3, 在保证LLZO 与活性物质良好的面接触的同时, 亦可实现扩散相的抑制.
固态电解质材料较高的杨氏模量一定程度上有助于阻挡锂枝晶生长但其缺点除无法直接通过加压的方法制备全固态电池外, 还会面临充放电过程中因为活性物质体积应变而带来的界面接触的恶化(图6(a)). 以钴酸锂为例[83], 其充电过程中存在a轴变短而c轴变长的趋势, 产生较明显的晶格畸变, 而放电后晶格恢复原状, 因此造成原本牢固的面接触破裂与粉化, 导致界面接触恶化, 增大界面阻抗, 加速循环衰减. 为解决正极活性物质因体积应变而带来的界面问题, 本课题组尝试采用了具备较低杨氏模量的反钙钛矿电解质作为助烧结剂,同时利用其较低的熔点(267 ℃)以避免较高的烧结温度、较高的离子电导率(6.8 × 10—5S·cm—1)以提供快速的锂离子传输通道, 通过热压共烧结方法获得界面稳定的力学性能, 实现全固态电池的稳定循环[70]. 在负极界面, 金属锂剥离而产生的间隙也会产生高的界面阻抗. 组装后的电池由于外加压力有限以及氧化物电解质较大的硬度, 放电后因金属锂剥离而产生的间隙无法被外压吸收, 导致产生了远高于充电态的负极界面阻抗(图6(b)). 采用柔性中间层可有效缓解因锂的沉积-剥离而产生的界面应力, 实现负极界面的力学稳定. Shao 等[73]采用铅笔涂画的方式在LLZO 表面引入具有较低杨氏模量的石墨中间层, Li/LLZO/Li 对称电池获得了1000 h 以上的稳定循环. 此外, 通过对施加压力的调控, 在大于临界组装压力条件下, 同样可以有效地减小放电后因金属锂剥离而产生的间隙[84].

表2 常见固态电解质、正极材料以及界面修饰层的杨氏模量Table 2. The Young’s modulus of the conventional solid-state electrolytes, cathodes and interface modification layers.
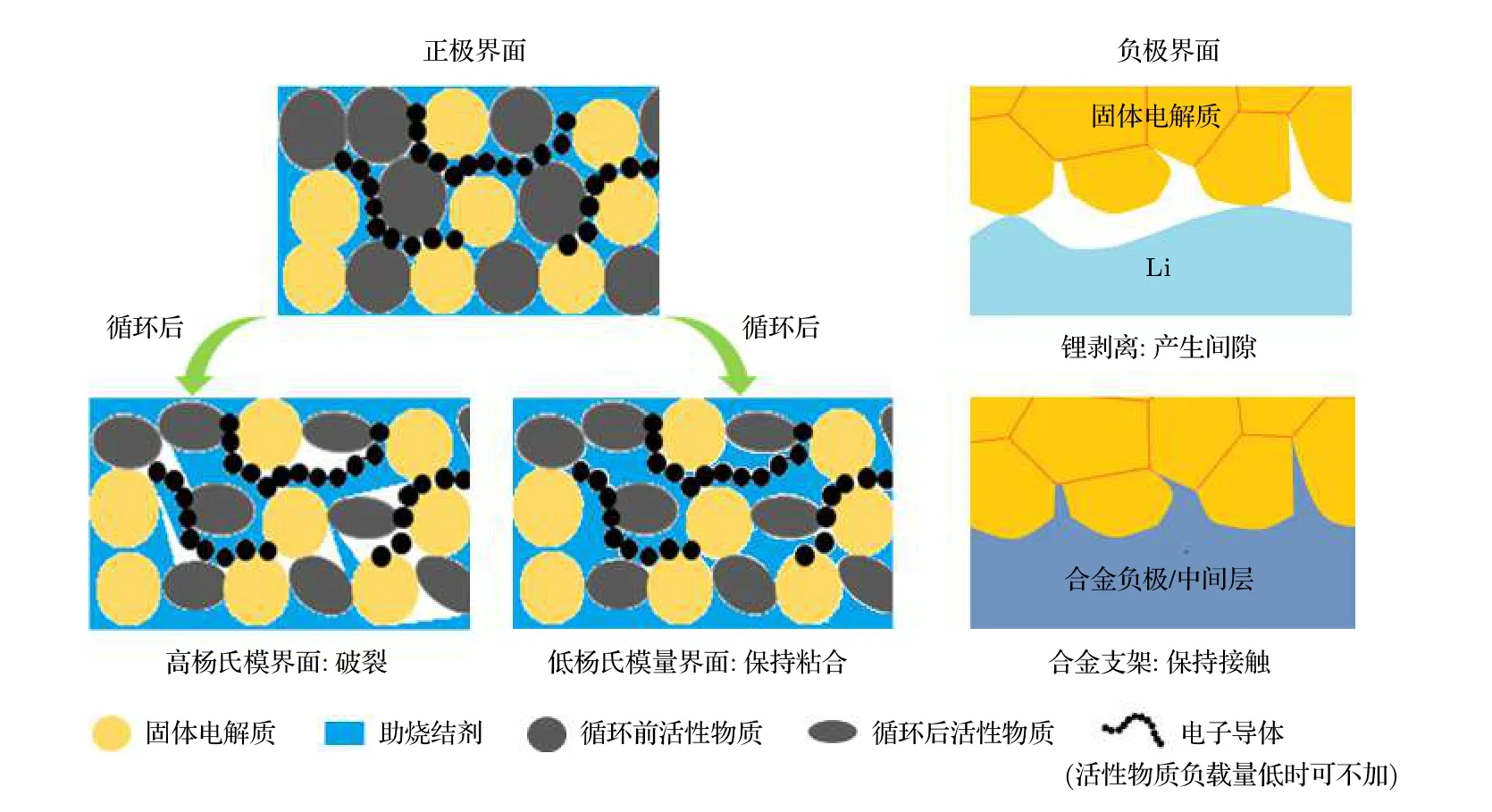
图6 (a) 活性物质循环过程中体积应变对正极界面接触的影响以及低杨氏模量中间层维持界面牢固接触示意图; (b) 负极界面锂剥离态导致间隙的产生以及加压或合金支架维持界面接触示意图.Fig. 6. (a) Schematic diagram of the effect of volume changes of the active materials during charge/discharge on the contact of cathode interface, and solid contact maintenance by low Young's modulus interlayer; (b) schematic diagram of the gap generated by Li stripping and solid contact maintenance by pressure or alloy frameworks.
5 提高界面稳定性的策略
5.1 界面稳定性的预测
第一性原理计算以量子力学为基础, 无需任何经验参数, 即可对多种材料性质作出预测. 这种原子尺度的模拟计算, 一方面能为新材料的机制机理提供深入的理解, 另一方面能为新材料的研发和改进提供指向性的设计策略. 在材料基因组的大背景下, 第一性原理计算在电池领域包括预测反应路径、电极电位、以及界面稳定性等方面大显身手.基于材料基因组的高通量计算, 能快速涵盖大量材料, 适合从大量数据中提炼总结规律, 从而对新材料的设计提供指导[16,85]. 在界面稳定性预测方面,主要通过热力学计算构建稳定的多元相空间, 在此基础上计算被研究相在不同的环境下与热力学稳定相的能量差, 从而确定其在该环境下的稳定性、热力学反应路径及其相应的反应电位. 值得一提的是, 这种基于热力学稳定相的计算, 忽略了化学反应动力学对电化学反应的影响. 有些电化学反应在反应动力学的限制下, 通过动力学更优的亚稳态进行反应甚至部分反应无法发生.
5.2 负极界面的构建
锂金属/负极界面: 在已知的常见固态电解质中, 少数材料不含金属阳离子, 因此具有对锂金属的兼容性(如LiPON, Li-P-S 体系材料). 其他固态电解质中, 金属阳离子往往会被锂金属还原. 还原反应生成包含金属或合金态的界面层产物, 从而形成离子电子混合导体的界面, 并进一步导致电池性能降低乃至失效. 解决策略之一是采用涂层的方式, 使用与锂金属兼容的薄层, 即类似人工SEI(solid electrolyte interphase)膜来实现钝化界面. 解决策略之二是通过在界面引入高浓度氮或氟掺杂, 从而导致包含氮化物或氟化物的钝化界面层的生成, 热力学计算(图7)发现, 很多金属氮化物和氟化物对锂金属枝晶具有抑止作用. 所以如果金属阳离子的使用不可避免, 使用氮化物和氟化物能实现对金属阳离子还原的保护, 从本质上解决这个问题. 界面疏锂是解决锂枝晶的关键.
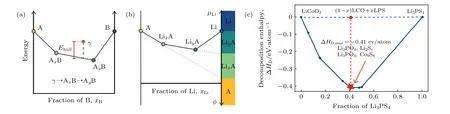
图7 基于材料数据库的热力学计算 (a)相稳定性: 被研究的亚稳态γ 相能量与同成分下热力学平衡相的能量差(energy above hull)是衡量γ 相稳定性的重要指标之一; (b) 巨电势相图(grand potential phase diagram): 衡量相稳定性在不同环境(比如对锂电位)下的变化; (c) 界面稳定性: 两相在不同比例时的二元相图及其相应的热力学反应焓变Fig. 7. Schematic illustrations of thermodynamic calculations: (a) Schematic of an energy convex hull, indicating the energy above hull Ehull of a metastable γ phase and its decomposition reaction into the phase equilibria; (b) schematic of a GPPD, illustrating the evolution of phase equilibria under changing Li chemical potential mLi and an applied voltage 4; (c) mutual reaction energy versus composition of a pseudo-binary composed of LiCoO2 and Li3PS4. The star corresponds to the predicted phase equilibria with decomposition enthalpy DHD at the mixing ratio.
5.3 正极界面的构建
在界面处使用氧化物涂层, 可以阻隔硫系电解质与电极的接触, 从而解决界面反应造成的化学不兼容性. 氧化物涂层材料通常具有较宽的电化学窗口, 从而可以极大改善本不兼容的硫化物电解质-正极界面, 计算筛选为涂层材料的选择提供了快速准确的指导. 目前常用的涂层包括LiNbO3[86],Li3PO4[87], Li3BO3-Li2CO3[88], Al2O3[89], Li3InCl6[90]等. Jung 等[88]使用Li3BO3-Li2CO3作为保护涂层包覆LiCoO2, 不仅可以抑制LiCoO2与Li6PS5Cl之间的化学反应, 还可以减小它们之间的空间电荷层, 恒电流充放电与恒电流间歇滴定技术均表明包覆层显著减小了电池的极化并增大了电池的充放电容量. 此外, 还可以使用原子层沉积法/激光脉冲沉积法在活性物质上包覆固体电解质从而增强两者之间的接触, 并提高电池的能量密度[91,92]. 例如, Wang 等[91]在已经包覆了LiNbO3的LiCoO2上使用原子层沉积法进一步包覆上Li10GeP2S12,从而最小化两者之间的空隙, 减小正极处的界面阻抗, 使全固态电池拥有更小的极化与更加优秀的电化学性能. 而根据Zhang 等[59]的研究结果, 导电碳的存在实际上是对正极界面稳定性有害的, 因此应对导电碳的使用谨慎对待. Tan 等[93]的研究结果表明, 可以通过调整碳的形态与比表面积来控制电解质分解的动力学. 在表面积减小的情况下, 固体电解质将更小地暴露于导电表面, 如果没有足够的电子途径发生氧化还原反应, 电解质分解的动力学则会大大降低. 因此, 使用纳米碳纤维(24 m2/g)替代全固态电池中经常使用的乙炔黑(80 m2/g)可以有效抑制电解质的电化学分解. 针对氧化物基电解质与正极较差的物理兼容性, 可引入助烧结剂通过高温烧结增加复合正极致密度, 助烧结剂要求同时兼具: 1)较高的氧化电位以保持其化学稳定性; 2)较高的离子电导率以提供锂离子的快速迁移通道; 3)较低的熔点以保证氧化物电解质与活性物质的黏连以实现充分接触, 并将其充分隔绝以防扩散相的生成; 4)较低的杨氏模量以缓冲活性物质充放过程中的体积应变.
6 总结和展望
基于全固态电解质的固态锂电池已经成为电池领域最具潜力的技术方向, 然而其电化学稳定窗口窄的问题渐渐得到学术界的正式和重视. 在未来的研究中, 解析电解质在电化学过程中的分解反应有望帮助我们了解其限制因素, 通过元素掺杂等方法可以适当拓宽电化学窗口. 除此之外, 从多层复合电解质的构建入手, 使正极侧电解质抗氧化性增强, 负极侧电解质抗还原性增强, 亦具有实际意义.在全固态电池的构建中, 形成动力学稳定的钝化界面, 是保证高电压正极和锂金属负极能够使用的关键. 为了有效抑制锂枝晶生长, 构建兼具高的疏锂界面能和高机械强度的界面层, 将是一个很有潜力的方向. 除此之外, 在全固态电池迈向实用化的过程中, 界面的力学稳定性需得到更多的关注, 力学稳定性问题则主要体现在电池循环过程中电解质与电极的接触损失, 为避免或抑制接触损失, 一方面将全固态电池在大于临界组装压力下运行以保持放电态金属锂与电解质的牢固接触; 另一方面,需考虑杨氏模量的影响, 采用低杨氏模量电解质或电极材料, 或引入低杨氏模量缓冲层, 以保持界面的力学稳定性.
猜你喜欢
卫星电视与宽带多媒体(2022年12期)2022-10-27
卫星电视与宽带多媒体(2022年18期)2022-10-26
消费电子(2022年6期)2022-08-25
中学生数理化(高中版.高考理化)(2022年5期)2022-06-01
中学生数理化(高中版.高考理化)(2021年5期)2021-07-16
陶瓷学报(2021年1期)2021-04-13
陶瓷学报(2021年1期)2021-04-13
消费导刊(2019年5期)2019-08-22
电子制作(2017年6期)2017-12-30
电子制作(2017年8期)2017-06-05
