
水杨醛席夫碱配体在聚合反应中的研究进展
2020-12-04 05:04孔勇
科学技术与工程 2020年30期
孔 勇
(中国石化石油工程技术研究院,北京 100101)
20世纪90年代以来,非茂金属催化剂引起了广泛关注。Fujita等率先报道了一种以水杨醛席夫碱为配体的前过渡金属聚合催化剂,其对烯烃聚合具有超高的催化活性[1-3]。随后,Wang等[4]、Mu等[5]将此配体推广到后过渡金属Ni、Pd体系。Younkin等[6]成功开发出中性镍催化剂,不需加入助催化剂即可常压催化乙烯聚合和乙烯/降冰片烯衍生物的共聚反应。水杨醛席夫碱配体由于高度稳定的骨架结构和良好的耐受性,广泛地应用于多种聚合反应,成为聚合催化剂的通用配体[7-9]。
首先简要介绍水杨醛席夫碱配体的种类,重点介绍配体金属化方法,并对其在烯烃配位聚合、CO2聚合、内酯开环聚合、原子转移自由基聚合和开环易位聚合等聚合反应中的应用进行评述。
1 水杨醛席夫碱配体的种类
水杨醛席夫碱配体可由水杨醛和伯胺通过缩合反应生成。水杨醛席夫碱按照配位数的不同可分为双齿 [O,N] 配体、三齿 [O,N,X] 配体和四齿 [O,N,N,O] 配体(图1)。其中,亚胺骨架可以是含氮的杂环化合物,如吡啶和咪唑等,目前研究最多的是邻酚氧基席夫碱 (77%)[10]。

图1 水杨醛席夫碱配体的种类Fig.1 Types of salicylaldehyde Schiff ligands
2 水杨醛席夫碱配体金属化
水杨醛席夫碱配体金属化一般是配体经NaH、BuLi、Et3N等碱负离子化,进一步与相应的金属盐反应或配体直接与相应的金属烃基化合物反应。目前水杨醛席夫碱配体过渡金属配合物几乎涵盖第3~14族的所有金属[11-13]。
Lara-Sanchez等[14]利用水杨醛席夫碱配体与M(CH2SiMe3)3(M=Sc、Y)在-20 ℃下反应得到预期Sc和Y的配合物,然而升温至室温则会得到三配体配位的产物(图2)。其在0 ℃下可高效催化己内酯的开环聚合,1 min内单体转化率达到100%。

图2 水杨醛席夫碱稀土金属配合物Fig.2 Rare earth metal complexes of salicylaldehyde Schiff ligands
Nakano等[15]利用Salen配体与CrCl2在空气中反应,不加入任何碱即可得到该配体的第六族金属Cr (IV) 的配合物(图3)。其在常压下催化环氧乙烷与CO2的交替共聚,所得聚合物为完全交替产物。
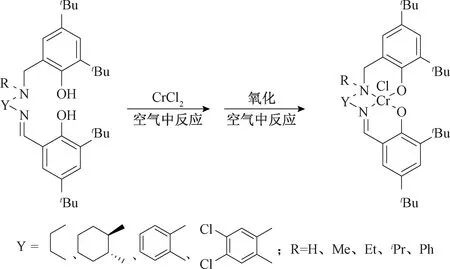
图3 Salen配体铬配合物Fig.3 Chromium complexes of Salen ligands
Qian等[16]利用(NH4)2Fe(SO4)2与配体在水/甲醇混合溶液中反应,生成Fe(Salphen)和Fe(Salen)化合物。在乙基铝氧烷(EAO)存在下,上述化合物可催化乙烯齐聚反应。水杨醛席夫碱配体钠盐与Fe2Cl4(thf)3反应可制得双配体配位产物。不分离水杨醛席夫碱配体利用一锅法反应,水杨醛、伯胺、Fe(OAc)2和KOH依次加入反应瓶同样可制得上述产物,在干燥的O2中会被氧化成含有O桥的双金属化合物(图4)[17]。

图4 水杨醛席夫碱铁配合物Fig.4 Iron complexes of salicylaldehyde Schiff ligands
Zhang等[18]利用水杨醛席夫碱的钠盐与trans-[NiCl(Ph)(PPh3)2]反应成功得到中性镍催化剂,但取代基位阻过大会阻止配体负离子化,利用此方法可制备其他双配体配位镍配合物和单配体配位产物(图5)。镍配合物可在助催化剂存在下或无须助催化剂催化乙烯均聚和共聚、其他烯烃均聚和共聚[19]。

图5 水杨醛席夫碱镍配合物Fig.5 Nickel complexes of salicylaldehyde Schiff ligands
目前几乎所有的过渡金属和主族金属水杨醛席夫碱配合物都已被报道。其中Ti配合物占据金属配合物总量的34%,Ni配合物为 25%,Zr配合物为 20%,金属V、Hf、Pd、Co、Cr配合物略低于 5%[20]。
3 烯烃配位聚合
烯烃配位聚合是目前水杨醛席夫碱配体应用最成功的领域。无论前过渡金属配合物还是后过渡金属配合物,甚至第13族的铝配合物和第14族的锡配合物均可催化烯烃配位聚合。最引人注目的催化剂是以第四族为代表的前过渡金属催化剂和以第十族为代表的后过渡金属催化剂。其中,FI催化剂又是上述催化剂中活性最高的催化剂,大量研究致力于FI催化剂的改良和扩展应用[21-22]。
三井化学(Mitsui Chemicals)的Fujita等成功开发出一系列水杨醛席夫碱第4族金属配合物,可高效催化烯烃配位聚合(图6)[23]。配体的取代基团、阴离子官能团都会对金属配合物的立体结构产生影响,丰富的立体结构会对聚烯烃的规整度产生重大影响[24]。在催化乙烯聚合中,配体取代基团(R1、Rn和R2)和金属的种类都会对聚合活性和聚合物的分子量产生重大影响。在配体取代基团相同的情况下,不同金属的催化活性顺序为M=Zr≥Hf>Ti。增大配体上取代基团(R1、Rn和R2)的位阻能显著改变催化剂的催化活性和聚合物的分子量[25]。

图6 FI催化剂Fig.6 FI catalysts
其中,Rn为甲氧基,金属原子为Zr,R1为环己基,R2为大位阻的异丙基苯基,催化乙烯聚合的活性高达8×106g/(mol·h·kPa)。该催化剂不仅可以催化乙烯聚合,还能催化其他烯烃聚合。例如当金属原子为Ti,以iBu3Al/[CPh3][B(C6F5)4]为助催化剂,催化1-己烯聚合得到无规、高分子量的聚1-己烯。此外,还可催化丙烯和乙烯的活性聚合,制备高度间规聚丙烯[26]。
Hu等[27]和Wang等[28]利用三齿[O,N,X]配体,成功制得其Ti配合物(图7)。配体边臂引入P、S、Se杂原子,提高了催化剂的稳定性,以MMAO为助催化剂,上述Ti配合物可高效催化乙烯聚合和乙烯与其他α-烯烃共聚[29]。即使Al/Ti降低至50,催化剂依旧对乙烯聚合具有超高聚合活性,活性可达到105g PE / [(mol of cat.) h kPa][30]。此外,上述催化剂不仅可催化乙烯与α-烯烃共聚,同样可以催化乙烯与极性单体共聚,极性单体插入率达到10%[31]。

图7 水杨醛席夫碱配体钛配合物Fig.7 Titanium complexes of salicylaldehyde Schiff ligands
Waltman等[32]将水杨醛席夫碱配体引入到后过渡金属催化剂的合成。此类催化剂与前过渡金属催化剂相比具有更好的基团耐受性,对聚合反应的条件要求更加温和。其中,中性镍催化剂由于卓越的催化性能引起了人们广泛的关注 (图8)[33-34]。中性镍催化剂不需要加入MAO、AlMe3、B(C6F5)3等助催化剂即可催化乙烯和降冰片烯等α-烯烃的均聚和共聚[35]。配体上的取代基团同样能够影响聚合活性、聚合物的分子量和分子量分布。Ni-Me化合物易发生烷烃消除反应,催化剂则会失活并生成双配体配位的镍配合物。由于后过渡金属镍催化剂的高度稳定性和对极性官能团的耐受性,上述催化剂不仅可催化乙烯齐聚、极性单体均聚和共聚,而且可以应用于水相和超临界CO2催化烯烃聚合[36]。

图8 中性镍催化剂Fig.8 Neutral nickel catalysts
除前过渡金属Ti、Zr和后过渡金属Ni配合物外,稀土配合物、铁配合物、钒配合物、铬配合物、钴配合物、铝配合物和锡配合物同样可催化烯烃配位聚合。其中,V配合物在室温下催化乙烯聚合,活性为223 g PE/ (mmol·V ·h ·kPa),所得聚合物重均分子量大于100 kg /mol,升温至70 ℃则催化活性降低,聚合物重均分子量降至30 kg/mol以下[37]。双配体配位的V配合物具有很高的热稳定性,以MAO为助催化剂,在75 ℃乙烯聚合活性可达到65 100 kgPE/ (mol·cat·h)[38]。单茂Cr配合物以AlMe3为助催化剂,即使在Al/Cr降至25依旧可以高效地催化乙烯聚合,所得聚合物具有很高的分子量 (1.00×106~1.45×106g/mol)[39]。
尽管单活性中心催化剂可以控制烯烃聚合物的分子量和聚合物的规整度,但最近研究发现有机反应中多个活性中心能够相互协同作用,提高催化聚合反应效率和选择性。Salata等[40]利用四齿 [N,O,O,N] 配体分别报道了双金属Zr和Ti催化剂 (图9)。刚性、平面的配体骨架可以有效防止两个金属原子在催化聚合过程中旋转、远离,从而减弱相互的协同作用。与单活性中心催化剂相比,双金属催化剂在MAO活化下可以制备线性高分子量聚乙烯,催化活性大约是相应单金属催化剂的8倍[41]。双金属催化剂催化乙烯与己烯、乙烯与1-辛烯的共聚,与相应单金属催化剂相比,α-烯烃在共聚物中的含量提高1倍。此外,上述催化剂还可催化乙烯与亚甲基环烷烃[亚甲基环戊烷(MCP)、亚甲基环己烷(MCH)],α,ω-二烯(1,5-己二烯、1,4-戊二烯)的共聚[42]。

图9 双金属Zr和Ti催化剂Fig.9 Bimetal Zr and Ti catalysts
中性镍催化剂的卓越催化性能引起了人们广泛关注,化学家陆续开发了基于水杨醛席夫碱配体的双核中性镍配合物 (图10)[43-44]。与单核镍催化剂相比双核中性镍催化剂具有更高的催化活性,制得的聚乙烯具有更高的分子量[45]。单体配位插入提高了活性中心的空间位阻和电子效应,从而提高了催化活性[46]。双核催化剂还可催化乙烯与降冰片烯衍生物、丙烯酸酯类的共聚,极性单体单元在共聚物中含量达到10%[47]。

图10 双核中性镍催化剂Fig.10 Dinuclear neutral nickel catalysts
Malgas-Enus等[48]报道了树枝状配体,并制备了多核镍催化剂(图11)。以MAO为助催化剂,双核催化剂和四核催化剂都可以催化降冰片烯聚合,四核催化剂的催化活性高于双核镍催化剂。

图11 多核镍催化剂Fig.11 Polynuclear nickel catalysts
Champouret等[49]利用吡啶水杨醛席夫碱配体制备了Fe、Co、Ni、Zn的双核金属配合物。加入过量MAO,Ni和Co的双核金属配合物可催化乙烯齐聚。
4 CO2聚合
CO2与环氧化物制备聚碳酸酯 (PC) 是CO2利用的一个热门研究方向。目前基于水杨醛席夫碱骨架的过渡金属催化剂主要为Salen配体的铬、钴和铝配合物[50-51]。
Darensbourg等[52-53]受Jacobsen利用Salen Cr配合物不对称催化环氧烷烃开环反应的启发,研究了Salen Cr配合物催化CO2与环氧化物共聚性能(图12)。但催化剂需加入N-甲基咪唑、[PPN]N3等亲核试剂作为助催化剂活化,催化环氧己烷 (CHO) 或环氧丙烷 (PO)与CO2共聚,转化频率(TOF)达到192 h-1,共聚选择性达到93%和碳酸酯连接单元含量为99%,分子量为13 000~26 000,分子量分布为1.10[54]。Rao等[55]报道了基于Salen骨架配体的Cr配合物,以DMAP为助催化剂催化PO和CO2共聚,在30 min内即可高效催化PO和CO2共聚。Nakano等[56]报道了Salalen配体的Cr配合物,加入1当量的[PPN]Cl,转换效率(TOF)达到230 h-1,所得聚合物的分子量为8 000,分子量分布为1.10~1.15。

图12 Salen Cr催化剂Fig.12 Chromium catalysts of Salen ligands
Qin等[57]报道了Salen Co化合物催化聚碳酸酯的制备,共聚选择性大于 99%,碳酸酯连接单元达到99%,TOFs为70 h-1。加入[PPN]Cl做助催化剂,催化rac-PO与CO2共聚,TOF达到620 h-1,共聚物的分子量为26 800,分子量分布为1.13[58]。随后,Lü等[59]陆续报道了其他Salen Co催化剂(图13)。Niu等[60]系统研究了引发基团X (Cl、Br、NO3、ClO4) 和助催化剂(R4NX、[PPN]X、MeIm、MTBD) 对Salen CoX催化PO与CO2共聚的影响。催化PO和CO2共聚,TOF最大可达到501 h-1(60 ℃、2.02×106Pa),共聚物分子量为5 400,分子量分布为1.10。延长反应时间和改为温和的反应条件,分子量最高达到7 000,TOF降至5 h-1,共聚选择性大于 99%[61]。

图13 Salen Co 催化剂Fig.13 Cobalt catalysts of Salen ligands
Nakano等[62]、Noh等[63]报道了新型Salen Co催化剂,该催化剂含有哌啶和哌啶盐两个边臂。质子化的哌啶盐边臂可通过质子化共聚物链阻止环碳酸酯的生成,开创了合成单组分Salen Co催化剂的新时代。通过在Salen配体边臂引入可替代助催化剂的官能团,提高催化剂的活性和选择性 (图14)[64]。基于此思想,在配体骨架边臂上引入季铵盐,使得游离、不断增长的共聚物阴离子链更加靠近活性中心[65]。这种设计使得Salen Co与边臂官能团存在比较强的分子内相互作用,提高了催化剂的催化活性和热稳定性。催化PO和CO2共聚,反应温度为80 ℃,2 020 KPa下,催化剂与单体比1∶50 000,转化数(TON)可达到14 500 h-1,TOF高达到3 200 h-1,得到的共聚物分子量为53 000,共聚选择性大于 99%,分子量分布为1.35[66]。与多组分催化体系相比,单组分催化剂用量大幅度降低,制得聚合物分子量更高,最大可达到95 000,而双组分催化体系最大分子量为30 000[67]。随后,化学家在Salen骨架上引入叔胺或Lewis碱作为助催化剂,合成了其他新型催化剂,催化PO制备聚碳酸酯(PPC),TOF可达到10 880 h-1,共聚选择性为97%[68]。
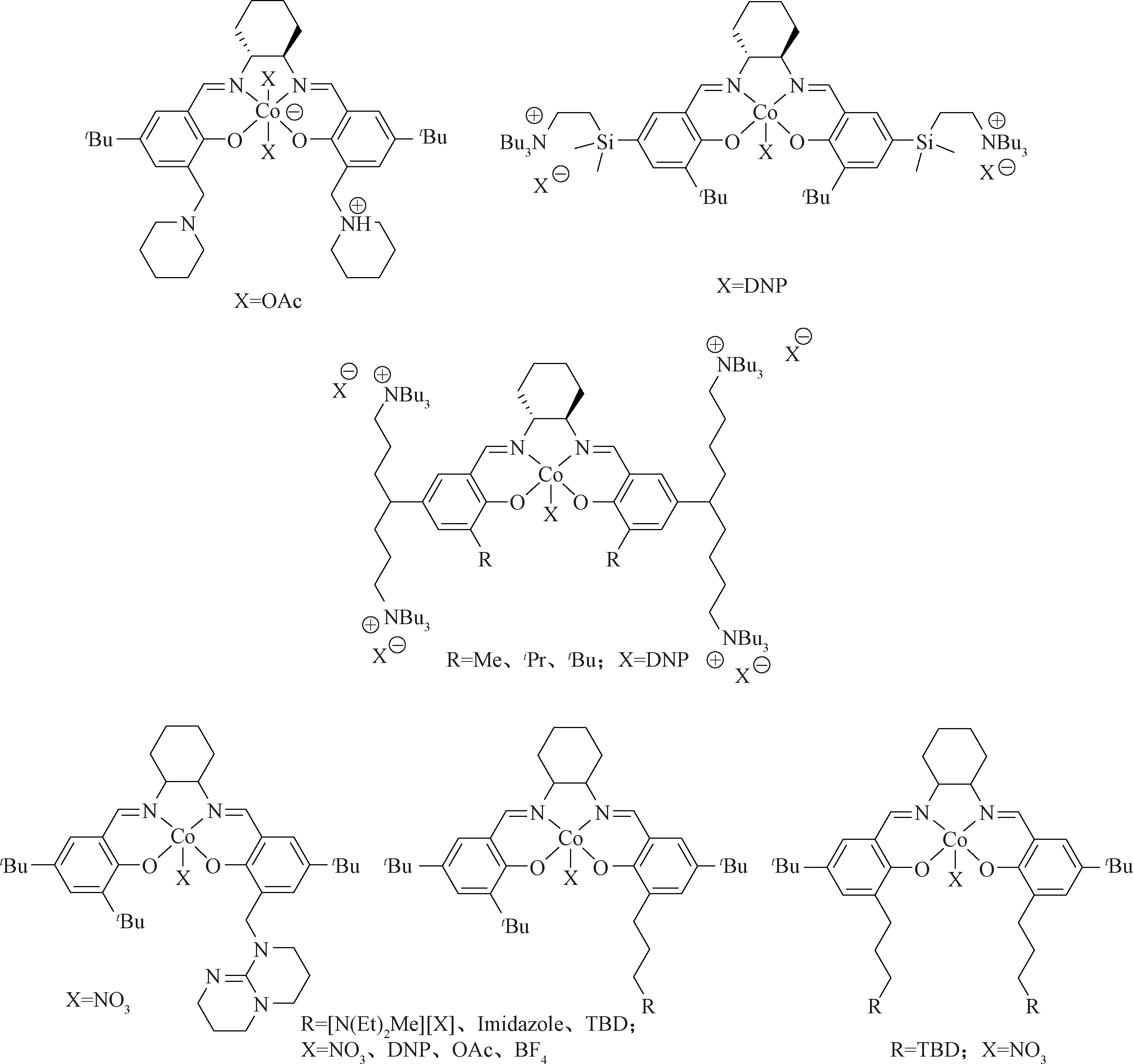
图14 单组分Salen Co催化剂Fig.14 Single component cobalt catalysts of Salen ligands
Salen的其他金属配合物同样可催化CO2和环氧烷烃的共聚反应。Darensbourg等[69]报道了一系列Al配合物,以季铵盐 (nBu4NX,X = Cl、Oac、N3) 或Lewis碱 (DMAP、N-MeIm、吡啶) 为助催化剂,催化CHO和CO2共聚80 ℃,TOFs为 5 ~ 35 h-1。但Salen Fe(III)、Zn、Ga、Mn(III) 配合物则不能催化CO2和环氧烷烃共聚,或只能催化环碳酸酯的合成[70]。
5 内酯开环聚合
基于水杨醛席夫碱配体的过渡金属配合物不仅可高效催化烯烃的配位聚合,而且还可催化ε-己内酯、丙交酯等内酯的活性开环聚合。所用的金属催化剂一般是第3族的稀土金属、第12族的Zn、第13族的Al和第14族的Sn。
Ovitt等[71]利用Salen骨架配体合成了Y配合物,尽管该配合物为二聚结构,但同等条件下其活性高于相应的Al配合物 (图15)。Chisholm等[72]利用大位阻的水杨醛席夫碱配体合成了Zn配合物,室温下以苯为溶剂,可催化内酯开环聚合。
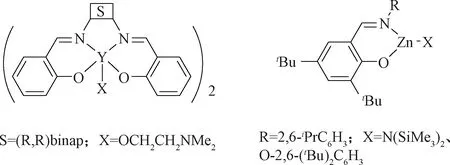
图15 内酯开环聚合催化剂Fig.15 Saturated N-heterocyclic carbene and unsaturated N-heterocyclic carbene
具有卟啉类似骨架的Salen配体与AlR3或Al(OR)3反应生成Al配合物(图16)[73-74]。上述催化剂均可以在标准条件下催化内酯开环聚合,没有任何诱导时间,单体转化率达到100%。此类配合物催化内酯开环聚合均可很好的控制聚合反应,所得到的聚合物分子量分布窄(<1.1),分子量与转化率成线性关系,具有手性结构骨架的配合物还可制备等规聚内酯[75]。

图16 Salen Al催化剂Fig.16 Aluminum catalysts of Salen ligands
6 原子转移自由基聚合
Monsaert等[76]报道了水杨醛席夫碱Fe配合物应用于原子转移自由基聚合(ATRP)研究 (图17)。Gibson首先合成了Fe(III) 配合物,研究了其催化苯乙烯和甲基丙烯酸甲酯(MMA)的原子转移自由基聚合,但是可能由于反应过程中生成Fe(II) 三配位的副产物导致聚合失控[77]。利用三齿配体合成Fe(II) 配合物,则可高活性地催化苯乙烯原子转移自由基聚合,分子量分布只有1.07[78]。

图17 水杨醛席夫碱铁催化剂Fig.17 Ironcatalysts of salicylaldehyde Schiff ligands
Clereq等[79]将水杨醛席夫碱配体引入到Grubbs类型催化剂的合成 (图18),该类型催化剂可催化苯乙烯、丙烯酸酯类的原子转移自由基聚合。将PCy3替换为氮杂环卡宾得到配合物,催化甲基丙烯酸甲酯原子转移自由基聚合,大幅提高甲基丙烯酸甲酯的转化率,分子量分布为1.2~1.3。

图18 水杨醛席夫碱引入Grubbs催化剂Fig.18 The introduction of salicylaldehyde Schiff ligands into Grubbs catalysts
7 开环易位聚合

Clercq等[83]、Drozdzak等[84]利用 [(pcymene)RuCl2]2与水杨醛席夫碱配体反应成功制得Ru系配合物 (图19)。不加入N2CH2SiMe3(TMSD),催化降冰片烯开环易位聚合,单体转化率只有 6%,即使加入TMSD作为引发剂,产率也只能达到35% (85 ℃)。而将配合物中的Cl替换为C6F5,催化活性大幅度提高,不加入引发剂,催化降冰片烯开环易位聚合,单体转化率达到86%[85]。在催化体系中引入助催化剂Et2AlCl,则降冰片烯可以定量生成聚合物[86]。

图19 新型结构不明确Ru催化剂Fig.19 New ill-defined Ru catalysts
8 结论与展望
水杨醛席夫碱由于特殊的骨架结构和优异的催化性能,广泛应用于烯烃配位聚合、CO2聚合、内酯开环聚合、原子转移自由基聚合和开环易位聚合等聚合反应中。在烯烃配位聚合、CO2聚合和内酯开环聚合中,水杨醛席夫碱金属配合物表现出优异的催化性能,但在原子转移自由基聚合和开环易位聚合中,其催化活性和选择性尚需进一步开展相关研究。未来水杨醛席夫碱聚合反应研究应主要集中在以下两点。
(1)负载型聚合催化剂研究。目前聚合研究主要集中在均相催化研究,聚合催化剂进行复载及回收研究较少,亟需加强催化剂负载和回收利用研究,推动其在聚合工业中的应用。
(2)新型聚合催化剂研究。目前水杨醛席夫碱配体在原子转移自由基聚合和开环易位聚合中催化性能还需进一步研究,凾需通过设计新型水杨醛席夫碱配体结构,开发新型结构高活性金属催化剂。
猜你喜欢
炼油与化工(2022年4期)2022-10-10
电气电子教学学报(2022年3期)2022-07-30
燃料化学学报(2022年5期)2022-05-30
石油沥青(2022年2期)2022-05-23
科学家(2022年4期)2022-05-10
纺织科学研究(2021年7期)2021-08-14
新课程·上旬(2020年3期)2020-08-07
海峡科技与产业(2019年4期)2019-10-26
无线互联科技(2019年10期)2019-08-06
中国新技术新产品(2016年20期)2016-12-08
