
基于分子印迹技术分离淡竹叶中异荭草苷的研究*
2020-11-25 03:09:20朱俊访
世界科学技术-中医药现代化 2020年8期
朱俊访,李 博,聂 阳
(广东食品药品职业学院实验实训中心 广州 510520)
淡竹叶为禾本科植物淡竹叶(Lophatherum gracileBrongn)的干燥茎叶[1],具有解热、利尿、抗菌和抗肿瘤等药理作用[2]。淡竹叶在我国大部分地区均有分布,具有良好的药理活性且资源十分丰富。淡竹叶药材含有丰富的碳苷黄酮[3],包括牡荆苷、异牡荆苷、荭草苷、异荭草苷、日当药黄素和当药黄素等,其中以异荭草苷含量较高[4]。现有报道[5-6]表明,淡竹叶总黄酮具有清除自由基、抗氧化和抑制脂质过氧化反应等药理作用,其中荭草苷具有抗缺氧、保护心肌、抗血栓等作用,牡荆素具有抗心肌缺血等作用[7],异荭草苷具有较强的抗氧化作用[8]。淡竹叶生长周期短,更生能力旺,资源丰富,除药用外,尚可用于食品、保健品等方面,值得充分开发利用,因此,淡竹叶是一种具有广阔开发应用前景的天然药物。
传统的中药活性成分提取、纯化方法主要是先采用有机溶剂提取,如竹叶黄酮常用的提取方法有回流提取、超声提取等,然后通过层析、大孔吸附树脂、重结晶、制备色谱等进一步分离、纯化,存在步骤多而繁琐、溶剂消耗大、成本高、污染环境等方面的问题[9-11]。因此针对传统分离方法的缺点,有需要开发一种有效、简便、选择性高的分离新技术来解决这种高成本、低收率的问题,分子印迹技术恰好满足这一要求。分子印迹技术具有制备简单、成本廉价、能重复使用等优点,尤其是特异的分子识别性能、高效率的富集与分离能力能够使结构类似的物质很容易得到分离,使中药成分纯化工艺得以简化,节省了溶剂的用量,减少了环境污染,因此,分子印迹聚合物在中药活性成分的分离纯化应用中具有极大的发展空间[12-15]。
本研究利用分子印迹技术,以异荭草苷为模板分子,制备异荭草苷的分子印迹聚合物(MIPs),采用电镜扫描对MIPs 的结构进行了表征,对其吸附性能进行了评价,并研究了该聚合物对淡竹叶中黄酮类成分异荭草苷的吸附分离效果。分子印迹技术具有强特异性及选择性,在中药有效成分研究中具有良好的应用前景[16],通过本研究以期为分子印迹聚合物在中药有效成分分离上的应用提供一定的研究基础。
1 材料与方法
1.1 材料与仪器
本研究所用的材料与仪器包括:LC-2010A 高效液相色谱仪(日本岛津仪器公司);UV2600 紫外分光光度计(尤尼科仪器有限公司);JSM7800扫描电镜(日本电子株式会社);AUY120 电子天平(日本岛津仪器公司);HH-4 恒温水浴锅(常州澳华仪器有限公司);SB5200DTN 超声波清洗机(宁波新芝生物科技股份有限公司);异荭草苷对照品(质量分数99.26%,批号18020610),成都曼思特生物科技有限公司;淡竹叶(批号20160401,广州市华宇药业有限公司),经广东食品药品职业学院康大力副教授鉴定为淡竹叶;2-乙烯基吡啶(2-VP)、丙烯酰胺(AM)、偶氮二异丁氰(AIBN),科密欧化学试剂有限公司;乙二醇二甲基丙烯酸酯(EDMA),乔科化学有限公司;甲醇为色谱纯,其余试剂为分析纯。
1.2 功能单体选择及用量筛选
本研究采用紫外分光光度法考察异荭草苷与AM和2-VP 两种功能单体在甲醇中的相互作用,通过紫外光谱的变化,对功能单体进行筛选。在0.1 mmol·L-1异荭草苷溶液中分别加入不同浓度的AM 和2-VP,使异荭草苷与功能单体的比例分别为1∶2、1∶4、1∶6,充分振荡后放置12 h,分别以相同浓度的功能单体做参比,测定紫外光谱,分析紫外光谱变化,分析确定最佳功能单体的种类及配比。
为了确定模板分子与功能单体的最佳比例,固定制备工艺中除功能单体外的其他条件,通过改变功能单体的用量,分别制备印迹聚合物,根据所得聚合物的性状及平衡吸附量(Q)来判断功能单体最佳用量。
1.3 印迹聚合物的制备[17]
称取1.5 mmol 异荭草苷溶解于25 mL 甲醇/氯仿(2∶3)中,超声振荡10 min,再加入一定量的功能单体2-VP,超声振荡10 min静置30 min,加入30 mmol交联剂EDMA,加入50 mg 引发剂AIBN,充分混匀超声振荡10 min,通氮气15 min,密封,置于65℃水浴中聚合24 h,得果冻块状聚合物。将聚合物取出晾干后研磨,粉末过120目筛后放入索氏提取器中,用甲醇-冰醋酸(9∶1,V/V)回流提取,直至提取液在360 nm下无模板分子吸收,再用甲醇洗去残留的冰醋酸,粉末晾干后真空干燥24 h 后备用,得异荭草苷的分子印迹聚合物。空白印迹聚合物(NIPs)的制备除了不加模板分子异荭草苷外,其余步骤同MIPs的制备。使用扫描电镜测定所得异荭草苷MIPs和NIPs的外貌形态并进行分析。
1.4 印迹聚合物静态吸附性能实验
称取40 mg 的异荭草苷印迹聚合物10 份,分别放入25 mL具塞锥形瓶中,分别加入用甲醇配制的浓度为0.01 mmol·L-1、0.02 mmol·L-1、0.03 mmol·L-1、0.04 mmol·L-1、0.05 mmol·L-1、0.06 mmol·L-1、0.07 mmol·L-1、0.08 mmol·L-1、0.09 mmol·L-1和 0.10 mmol·L-1的异荭草苷对照品溶液,室温振荡24 h,取吸附后的溶液进行离心、过滤,滤液于360 nm 处测定吸光度。空白聚合物的静态吸附实验的处理和操作同上。根据吸附前后溶液中模板分子异荭草苷的浓度变化计算分子印迹聚合物的吸附量(Q),并绘制等温吸附曲线,计算公式见公式(1)。
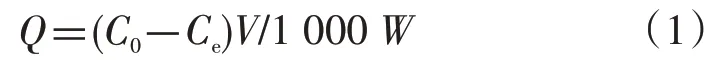
上式中Q为平衡吸附量(μmol/g),C0为溶液中异荭草苷的起始浓度(μmol/L),Ce为平衡时溶液中异荭草苷的浓度(μmol/L),V为溶液体积(mL),W为印迹聚合物的质量(g)。
1.5 动态吸附实验
取二支玻璃色谱柱分别加入2.5 g干燥的MIPs,得异荭草苷的MIPs 填充柱A 柱和B 柱,另外再取一玻璃色谱柱加入2.5 g NIPs 为C 柱,分别以甲醇平衡色谱柱。分别吸取浓度为0.10 mmol·L-1的异荭草苷对照品溶液0.5 mL 加至平衡后的萃取柱MIPs-A 柱、MIPs-B 柱和NIPs-C柱。MIPs-A柱先用甲醇进行淋洗,收集洗脱液,每份2 mL,再用甲醇-冰醋酸(9∶1)洗脱,收集洗脱液,每份2 mL。MIPs-B 柱直接用甲醇-冰醋酸(9∶1)洗脱,收集洗脱液,每份2 mL。NIPs-C柱用甲醇进行淋洗,收集洗脱液,每份2 mL。在360 nm 处分别测定各洗脱液吸光度,实验重复3次,分别绘制MIPs-A柱与MIPs-B柱、MIPs-A柱与NIPs-C柱的洗脱曲线。
1.6 淡竹叶提取液的制备
取淡竹叶100 g,粉碎后过50 目筛,使用80%乙醇浸泡提取24 h,提取3 次,合并提取液并浓缩至无醇味,使用等体积石油醚萃取3次,合并水相并浓缩得浸膏[18]。用甲醇超声溶解上述浸膏,过滤,定容至25 mL容量瓶中,得供试品溶液(含异荭草苷37.6%)。
1.7 HPLC分析
吸取淡竹叶提取液上样,用甲醇洗脱,分别收集MIPs 柱和NIPs 柱洗脱液。使用以下色谱条件进行分析,考察MIPs对淡竹叶中异荭草苷的分离效果。

表1 流动相梯度表
色谱条件:Agilent C18色谱柱(250 mm×4.6 mm,5 μm),流动相为甲醇和0.5%醋酸水溶液,按表1 进行梯度洗脱,进样量为10 μL,柱温25℃,体积流量为1.0 mL·min-1,检测波长为360 nm[19-20]。
2 结果与分析
2.1 功能单体选择
功能单体在聚合物制备过程中,主要是与模板分子通过共价或非共价作用形成复合物,模板分子和功能单体在聚合前能否形成稳定的复合物是获得高选择性分子印迹聚合物的关键,因此,选择合适功能单体种类及确定功能单体用量至关重要。增加功能单体的用量,会使模板分子和功能单体的预组装更完全,但用量过大单体自身会发生缔合,影响印迹聚合物中特异性结合位点的产生。本实验通过紫外-可见分光光度法来确定最佳功能单体的种类和配比。根据紫外光谱原理,价电子与氢原子形成氢键,电子能量发生变化,分子轨道发生扭曲变形,电子跃迁概率发生变化,这些原因导致吸光度发生变化。因此,通过比较紫外光谱的变化,可推测模板分子与功能单体间相互作用强度和配比等信息。
模板分子异荭草苷结构具有多个酚羟基,具有一定的酸性,羰基氧原子上的未共用电子对有微弱的碱性。理论上,选择碱性功能单体更有利于模板分子和功能单体形成稳定的复合物,因此以AM 和2-VP 为备选功能单体。使用紫外分光光度法分别对0.1 mmol·L-1异荭草苷、异荭草苷:AM(1∶2、1∶4、1∶6)的和异荭草苷:2-VP(1∶2、1∶4、1∶6)进行光谱扫描,结果见图1。可以看出异荭草苷与不同比例的2-VP 混合后,在250-290 nm 范围内的吸收曲线发生了明显变化,吸光值随2-VP 浓度增大而减小,说明模板分子与2-VP 之间有较强的作用力。异荭草苷与不同比例的AM 混合后,紫外图谱没有发生明显变化,说明模板分子与功能单体作用较弱。由此可推断2-VP 是一种较理想的功能单体,这可能是由于其与模板分子间存在氢键及酸碱作用,因此产生了较好的识别位点。
2.2 功能单体用量筛选
固定制备工艺中模板分子用量为1.5 mmol、交联剂的用量为30 mmol,将功能单体的用量分别设为3 mmol、6 mmol、9 mmol 和 12 mmol,分别制备印迹聚合物,记录所得聚合物的性状,计算平衡吸附量(Q)。结果见表2,不同的功能单体用量制备得到1-4 号MIPs,其中1-3 号聚合物为果冻状固体,而4 号聚合物由于添加的功能单体较多所形成的聚合物结构不够紧密因此为胶状。所以综合1-4 号MIPs 的性状和Q值可判断较佳工艺为3 号,即印迹分子异荭草苷的量为 1.5 mmol,功能单体 2-VP 的量为 9 mmol,交联剂EDMA的量为30 mmol。

图1 异荭草苷与不同浓度功能单体的紫外光谱
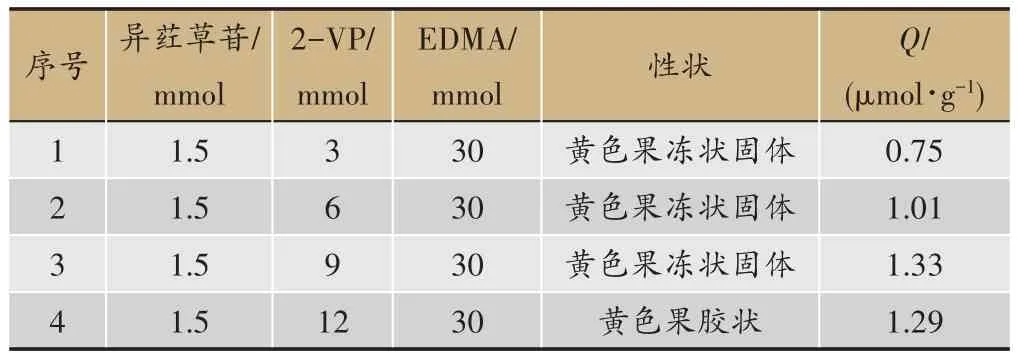
表2 功能单体用量筛选
2.3 聚合物表面形态研究
扫描电镜是一种微观形貌观察手段,主要是利用二次电子信号成像来观察样品的表面形态。使用扫描电镜对制备所得异荭草苷MIPs 和NIPs 的表面形态进行测定,研究聚合物的微观结构以及表面的形态差异,对聚合物的聚合情况进行评价。由图2可以看出,异荭草苷MIPs 和NIPs 的表面形态具有较大差异,NIPs 的表面较为光滑、均一、质地紧密,而异荭草苷MIPs 表面较为粗糙、具有大量的小孔,这可能是由于异荭草苷MIPs 中模板分子被洗脱后留下的“印迹”空穴,正是这些空穴使得该MIPs具有了特殊的吸附性和选择性,因此这更有利于对模板分子的吸附。
2.4 静态吸附实验
通过静态吸附实验研究异荭草苷在印迹聚合物和非印迹聚合物上的吸附行为,使用吸附量对平衡浓度作图,得吸附等温线见图3。由图3可见在所研究的浓度范围内,MIPs 对异荭草苷具有一定的吸附能力,吸附量随着溶液浓度的升高逐渐增大,最大吸附量可达 2.5 μmol∙g-1,在相同浓度下 NIPs 对异荭草苷的吸附相对较小。随浓度的增加,MIPs和NIPs吸附量之差越来越大,推测MIPs对目标分子具有选择性的吸附能力,而NIPs 不存在这种特异性的结合位点,且对模板分子只有弱的非选择性吸附,因此,NIPs 对目标分子吸附能力较差。

图2 分子印迹聚合物的SEM图片

图3 印迹聚合物的等温结合曲线
2.5 动态吸附实验
MIPs-A 柱、MIPs-B 柱和 NIPs-C 柱分别使用 0.10 mmol/L 异荭草苷对照品溶液上样,使用分光光度法分别测定异荭草苷在相同MIPs 柱不同洗脱液及MIPs 柱与NIPs 柱间的洗脱保留情况,以洗脱体积为横坐标,吸光值为纵坐标,绘制洗脱曲线。图4-①为MIPs 柱与NIPs柱上样后使用甲醇洗脱的泄露曲线图,由图可看出NIPs 柱比MIPs 柱更早出峰且峰型更窄,这是由于模板分子异荭草苷在MIPs中具有特异的结合位点,而在NIPs 中不具有这种结合位点,因此,异荭草苷在MIPs 柱上的保留时间更长。图4-②为MIPs 柱使用不同洗脱液洗脱的泄露曲线,MIPs-A 柱先使用甲醇洗脱、再用甲醇∶冰醋酸(9∶1)洗脱,相比MIPs-B 柱只使用甲醇进行洗脱,MIPs-A 柱在洗脱后期多出一个峰,这是由于MIPs 对模板分子异荭草苷具有一定的吸附能力,在使用更强的洗脱剂甲醇∶冰醋酸(9∶1)时被更集中的洗脱下来。通过对比动态吸附试验结果,可以说明制备所得MIPs 对模板分子异荭草苷具有一定的吸附能力且吸附能力比NIPs强。
2.6 方法学考察
2.6.1 线性关系考察
使用1.7 项下的色谱条件,测定异荭草苷的系列浓度的对照品标准溶液,平行测定3次,对平均峰面积(y)与质量浓度(x)进行线性分析,计算得回归方程y=251960x+251712(r=0.9991)。结果显示异荭草苷在17.44-52.32 μg/mL范围内线性关系良好。
2.6.2 精密度试验
精密吸取异荭草苷对照品溶液10 μL,采用1.7 项的色谱条件,连续进样6 次,测定异荭草苷峰面积,经计算RSD=0.22%(n=6),表明该方法精密度良好。
2.6.3 重现性试验
取同一供试品溶液5份,在1.7项的色谱条件进行测定,记录异荭草苷的峰面积,经计算RSD=2.38%(n=6),表明该方法重现性良好。
2.6.4 稳定性试验
取同一供试品溶液分别于0 h,2 h,4 h,8 h,16 h进样10 μL,记录异荭草苷的峰面积,经计算RSD=1.35%(n=6),结果表明供试品溶液在16 h内稳定。
2.6.5 回收率试验
采用加样回收法,精密称取已知含量的供试品6份,分别加入适量的异荭草苷对照品,按1.6 项操作制备供试品溶液,在1.7 项的色谱条件进行测定峰面积,计算异荭草苷的平均回收率为99.1%,RSD=1.40%。
2.7 淡竹叶中异荭草苷分离研究
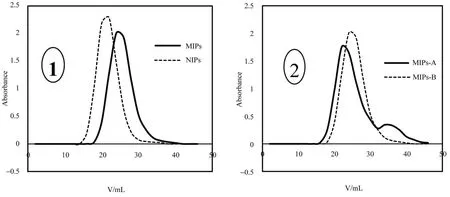
图4 异荭草苷的动态吸附曲线
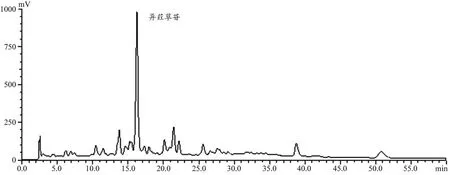
图5 淡竹叶提取液高效液相色谱图
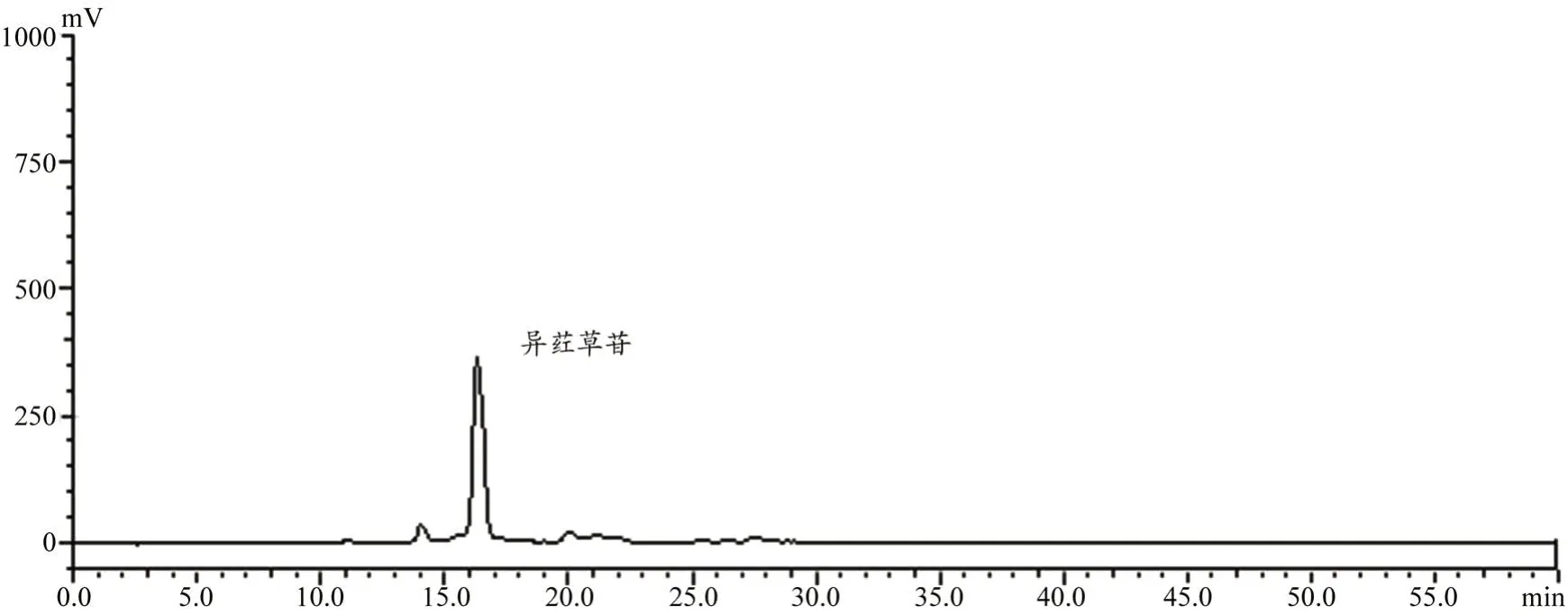
图6 淡竹叶洗脱液高效液相色谱图
淡竹叶中含有多种黄酮类化合物,其中以异荭草苷含量较高,图5 为淡竹叶提取液(含异荭草苷37.6%)使用1.7 项下色谱条件得到的高效液相色谱图,图6 为淡竹叶提取液经过MIPs 柱洗脱所得的中段洗脱液(含异荭草苷83.6%)在同一色谱条件下所得的高效液相色谱图。对比两色谱图可以看出,淡竹叶提取液经MIPs 柱洗脱后的中段洗脱液除异荭草苷外其他色谱峰明显减少,异荭草苷含量提高了46%,这可能是由于MIPs 对淡竹叶提取液中的异荭草苷具有一定的选择性吸附能力,而对淡竹叶中的其他成分没有选择性吸附。淡竹叶中的其他成分在MIPs 柱中的保留时间较短,主要在洗脱前期被洗脱下来,而异荭草苷保留时间较长洗脱较慢。因此,说明MIPs柱对淡竹叶提取液中的异荭草苷存在一定的纯化能力。
将淡竹叶提取液分别过MIPs 柱和NIPs 柱,收集洗脱液,洗脱液进行HPLC 分析,以异荭草苷为指标考察聚合物对异荭草苷的分离效果。以异荭草苷的百分含量为纵坐标,洗脱体积为横坐标,作异荭草苷在MIPs 柱和NIPs 柱的洗脱曲线图,见图7。通过比较两条曲线可以看出,由于MIPs柱对异荭草苷具有特异性吸附性能力,异荭草苷在MIPs 柱的保留时间要比在NIPs 柱长,因此可将该MIPs 用于异荭草苷的分离纯化。
3 讨论
本研究制备了异荭草苷分子印迹复合物,并对该聚合物的表面形态和吸附性能进行了研究,结果表明,该分子印迹复合物对异荭草苷有较好的结合性能与分离能力,这是由于MIPs与模板分子异荭草苷之间存在多个相互作用的位点和相互匹配的空间,且形成了氢键等非共价键,使得MIPs对模板分子具有较强的吸附能力。制备的分子印迹聚合物能有效的富集淡竹叶中的异荭草苷,质量分数可达83.6%,与传统方法相比,分子印迹聚合物在分离的效率、效果上有明显的提高,不过与制备色谱的分离结果相比[11],在纯度上仍存在一些差距。虽然制备色谱能达到很好的分离纯化效果,但也存在成本高、应用不便等问题。因此制备所得异荭草苷分子印迹聚合物的吸附性能还有待进一步提高,可考虑进一步优化制备条件、引入先进的制备方法,或者结合计算机模拟技术来提高合成和研发效率。
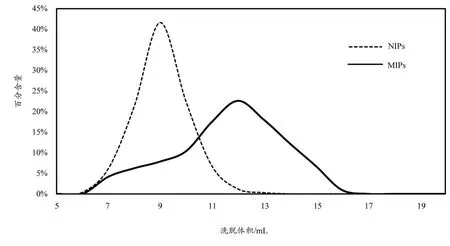
图7 淡竹叶中异荭草苷的洗脱曲线图
本研究为淡竹叶中异荭草苷的提取分离提供了新的方法,也为中药有效成分的分离纯化提供了新的思路。分子印迹技术具有制备过程简单、周期短、稳定性好、选择性好、消耗少等优点,应用于中药有效成分的分离纯化,可以提高分离效率,减少溶剂用量,并有助于推动绿色中药的发展。但是,分子印迹技术在其应用过程中仍然存在一些问题,如水相中分子印迹聚合物的不相容性、结合位点不均匀、模板渗漏等,因此,分子印迹技术在中药方面的应用有待更进一步的研究。
猜你喜欢
大众健康(2024年6期)2024-06-14 07:30:32
陶瓷研究(2022年3期)2022-08-19 07:15:18
云南画报(2021年10期)2021-11-24 01:06:56
吉林中医药(2021年5期)2021-03-27 14:31:30
中国粮油学报(2019年4期)2019-07-12 09:06:44
小学生优秀作文(高年级)(2018年4期)2018-09-11 01:23:22
中成药(2018年2期)2018-05-09 07:20:06
长寿(2017年9期)2017-07-20 23:04:41
中成药(2017年4期)2017-05-17 06:09:28
中老年健康(2015年9期)2015-05-30 23:22:10
