
微生物Rieske型芳香环双加氧酶研究进展*
2020-11-19 02:53龙丽娟
广西科学院学报 2020年3期
刘 玉,杨 键,李 茹,龙丽娟,3**
(1.中国科学院南海海洋研究所,中国科学院热带海洋生物资源与生态重点实验室,广东广州 510301;2.中国科学院大学,北京 100049;3.南方海洋科学与工程广东省实验室(广州),广东广州 511458)
0 引言
芳香族化合物是一类典型的有机污染物,在环境中分布广泛。该类化合物能够通过多种途径进入环境,且具有半衰期长、化学结构多样、毒性高等特点,能够被生物吸收富集并通过食物链传递,威胁人类健康与生态环境安全[1,2]。如何高效安全地消除这些有机污染物成为人们日益关注的焦点。环境微生物(细菌、真菌、藻类)能够依靠体内酶系统代谢天然或合成的芳香族化合物,是芳香族化合物生物地球化学循环的重要驱动者[3-6]。Rieske型芳香环双加氧酶能够催化芳香环特定位置的双羟基化反应,生成对应的顺式二氢二醇或二醇羧酸[7,8],从而降解芳香族化合物。有关Rieske型芳香环双加氧酶的研究多集中在新型双加氧酶的分离纯化[9]、晶体结构[10]、催化机制[11]、蛋白质工程[12,13]等方面,有效促进了该类酶在生物修复以及化学合成等领域的应用。该类酶具有催化化学反应多样性、高度立体选择性等特点,具有非生物催化剂无法比拟的优势,并在手性中间产物合成中发挥着巨大的作用,有望被开发为高效绿色催化剂[14]。
Rieske型芳香环双加氧酶能够催化包括羟基化、环氧化、去甲基化、去饱和、氯化等在内的多种化学反应[15],其生物学作用不仅仅局限于芳香族化合物的降解,而且还能参与一系列分解代谢与生物合成途径中的氧化转化。选择性氧化是催化领域的重要课题,目前基于重金属的催化剂既昂贵又不环保,因此寻找低廉环保、立体选择性和区域选择性高、催化高效的人工催化剂成为研究的热点。Rieske型芳香环双加氧酶本身具有天然的、无可比拟的高效催化能力,在开发能够氧化底物多样性且具有区域选择性和立体特异性的催化剂上拥有广阔的前景。为深入了解Rieske型芳香环双加氧酶的化学反应过程和选择性催化机理,本文对微生物Rieske型芳香环双加氧酶的分类、催化功能多样性、蛋白结构、催化机制以及蛋白质工程等方面的研究进行综述,期望为该类酶的进一步理论分析与实际应用提供借鉴。
1 Rieske型芳香环双加氧酶的分类
Rieske型芳香环双加氧酶是含有Rieske[2Fe-2S]中心的多组分酶系统,多数由末端氧化酶、还原酶和铁氧还原蛋白3部分组成[16]。其中,末端氧化酶由α亚基或αβ亚基共同组成[17],均含有一个Rieske[2Fe-2S]中心和一个单核铁活性位点。还原酶组分是一种含有黄素腺嘌呤二核苷酸(FAD)或黄素单核苷酸(FMN)的黄素蛋白,一些还原酶组分中同样含有[2Fe-2S]中心[7]。铁氧还原蛋白是含有[Fe-S]中心的小型酸性蛋白[8],根据[Fe-S]中心铁硫原子的连接数目可分为[2Fe-2S]和[3Fe-4S]两类[18]。最初,依据Rieske型芳香环双加氧酶的组分不同以及氧化还原中心性质差异,将双加氧酶分为三大类[16,19](表1)。然而随着更多新型加氧酶的发现,该静态分类系统存在着一定的局限性。Kweon等[18]基于多组分酶系统,将加氧酶作为一个整体进行分析,提出一个较为完善的新型动态分类系统。他们首先根据氧化酶的α亚基或αβ亚基组成将其分为两类,并根据保守区序列的不同将还原酶分为GR型、FNRC型、FNRN型3类,将铁氧还原蛋白分为[2Fe-2S]和[3Fe-4S]两类,最后综合以上多组分的类别,将加氧酶共分为五大类:Ⅰ类是由氧化酶和FNRC型还原酶组成的双组分系统;Ⅱ类是由氧化酶和FNRN型还原酶组成的其他双组分系统;Ⅲ类是由氧化酶、FNRN型还原酶和[2Fe-2S]型铁氧还原蛋白组成的三组分系统;Ⅳ类是由氧化酶、GR型还原酶和[2Fe-2S]型铁氧还原蛋白组成的三组分系统;Ⅴ类是由氧化酶、GR型还原酶和[3Fe-4S]型铁氧还原蛋白组成的三组分系统。该分类系统能够反映新的序列信息以及加氧酶组分之间的相互作用,可用于不完全的加氧酶分类。
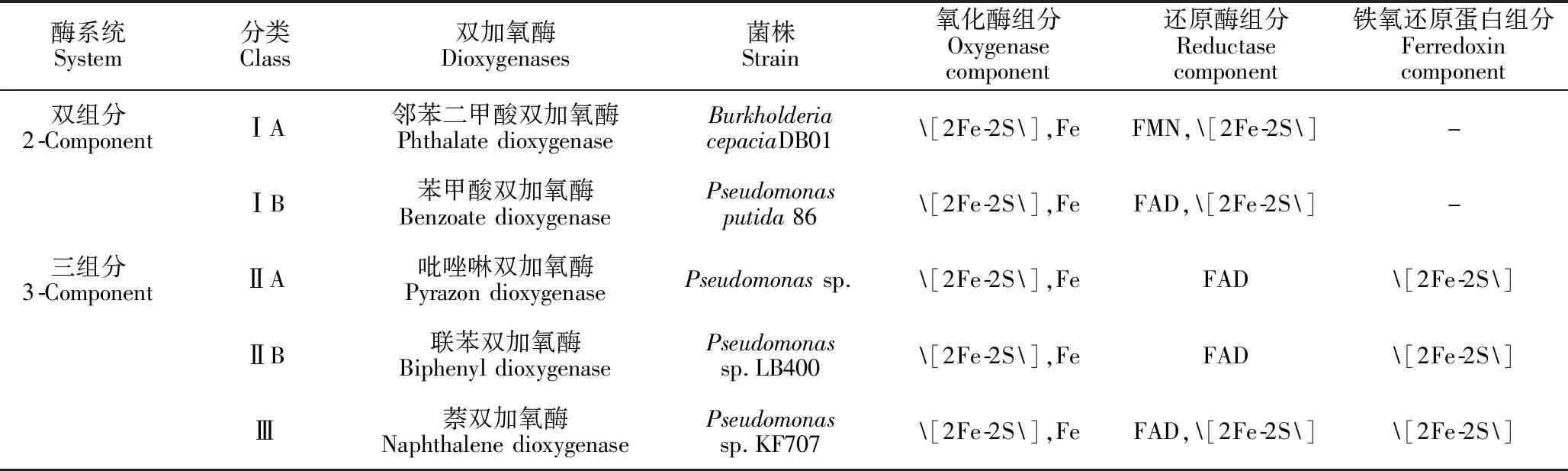
表1 Rieske型芳香环双加氧酶分类[16,19]Table 1 Classification of Rieske-type aromatic ring dioxygenases[16,19]
2 催化功能多样性
Rieske型芳香环双加氧酶能够催化多种反应,包括羟基化、环氧化、去饱和、氯化、脱烷基、去甲基化等(图1)[14]。例如,萘双加氧酶不仅能够催化多种双环和三环的芳香族化合物双羟化反应生成对应的二氢二醇,而且能催化其他氧化反应,包括单羟化、去饱和、脱烷基化和亚砜化等[20]。同属非血红素铁依赖型的Fe2+/α酮酸双加氧酶也能够以加氧的方式催化类黄酮进行环羟化、氧化、去饱和等反应[21](表2)。Rieske型芳香环双加氧酶具有宽泛的底物特异性,Baboshin等[22]研究认为芳香环羟化双加氧酶催化芳香烃化合物的特异性存在两个特征:1)同一的双加氧酶可催化不同的底物羟基化;2)对于不同的底物,同一双加氧酶表现的活性不同。计算分析结果显示,来自Sphingobiumsp.PNB的Rieske型芳香环双加氧酶具有降解多种芳香族化合物的能力,证明该加氧酶具有宽泛的底物特异性[10]。不同来源的联苯双加氧酶(BPDO)对同类底物的偏好和区域特异性存在显著差异,可以催化甲苯、多氯联苯、羟基联苯等底物[17,23]。来自PseudomonasKF707的联苯双加氧酶BPDOKF707不能催化联苯的3,4-位双羟基化,但对二氯联苯同系物却表现出不同的活性,较2,2′-或3,3′-二氯联苯而言,优先转化4,4′-二氯联苯[24]。另外,虽然BurkholderiaxenovoransLB400的联苯双加氧酶BPDOLB400与BPDOKF707的序列同源性超过95%,但BPDOLB400的转化范围更广。BPDOLB400能够优先转化含有多达6个氯的邻位取代同系物,也能催化2,2′,5,5′-四氯联苯3,4双羟基化,还能催化某些二氯同系物的脱卤作用,产生2,3-二羟基联苯。BPDOLB400与PandoraeapnomenusaB-356的联苯双加氧酶BPDOB-356均能催化联苯,但BPDOB-356具有更高的转化效率。联苯双加氧酶的α亚基(BphA1)直接影响酶的催化活性和底物特异性[25],该差异性是由α亚基上少量的氨基酸差异造成的[26]。BphA1活性位点中的氨基酸残基具有不同的作用,协同决定了酶的底物特异性[27]。β亚基对底物特异性的影响存在着两种不同的看法。Hurtubise等[28]通过对BPDOB-356和BPDOLB400的αβ亚基进行交叉重组实验,证明α和β亚基均影响酶与底物的相互作用,其中β亚基的结构不仅能影响酶对催化底物的氧结合能力,还能影响底物的反应模式。α和β亚基的结构特征决定了催化口袋的大小和形状,进而影响氧化底物的范围,如Sphingobiumsp.PNB的α亚基结构包含一个非常大的底物结合口袋,可以满足容纳高分子量底物的空间要求,因此能够催化多种芳香烃化合物[10]。而Kauppi等[29]研究工作表明α亚基是控制底物特异性的主要成分,β亚基似乎不参与底物的催化,可能仅作用于酶的结构稳定性。
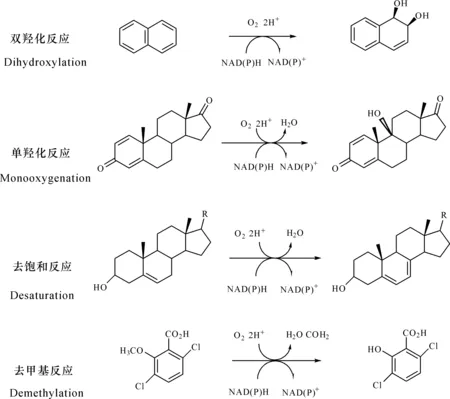
图1 Rieske型芳香环双加氧酶化学反应Fig.1 Chemical reaction of Rieske-type aromatic ring dioxygenases

表2 Rieske型加氧酶催化多样性Table 2 Diversity of Rieske-type oxygenases catalyzed reactions
3 蛋白结构
Kauppi等[29]和Furusawa等[27]通过X-射线衍射方式分别解析出来自Rhodococcus的萘双加氧酶和联苯双加氧酶的晶体结构。随后,来自Sphingomonas的环羟化双加氧酶和Sphingobium的联苯双加氧酶等的晶体结构也相继被表征[33-35]。在芳香环双加氧酶中,对萘和联苯双加氧酶的研究较为深入,而这两种酶的晶体结构也是最早被解析出来的,为其他双加氧酶的三维结构研究提供了参考。联苯双加氧酶采用α3β3构型,为六聚体结构,呈蘑菇状(图2a)。蘑菇的帽部由α亚基组成,茎部由β亚基组成。其整体结构以及各亚基的结构与萘双加氧酶(NDO)十分相似。每个αβ聚体通过一个非结晶的3倍轴与其他αβ聚体连接,且3个αβ聚体间没有明显的结构差异。SphingobiumyanoikuyaeB1的BPDOB1的α亚基由460个氨基酸残基组成,含两个与催化过程密切相关的结构域,分别为催化域和Rieske域,共同介导氧原子和电子转移[36]。BPDOB1催化域含有一个单核铁活性位点,单核铁与两个组氨酸(His)、一个天冬氨酸(Asp)及一个水分子(H2O)配位(图2b),组氨酸和天冬氨酸在酶中高度保守。BPDOB1Rieske域中两个硫化物离子桥接两个铁离子,形成扁平菱形排列,其结构与BPDO系统的铁氧还蛋白组分相似[27],包含一个Rieske[2Fe-2S]中心,并与两个半胱氨酸(Cys)和两个His配位结合(图2b)。BPDOB1中α亚基内的单核铁与Rieske[2Fe-2S]中心的距离约为40 Å(萘双加酶中两者的距离约为44 Å),而与相邻α亚基单核铁的距离约为12 Å,比同一个亚基内的距离短得多[17]。在酶反应过程中,电子由Rieske[2Fe-2S]中心传递至单核铁,距离特征表明电子更可能在α亚基之间发生转移。单核铁活性中心与相邻α亚基的Rieske[2Fe-2S]中心之间存在氢键,铁硫中心His121的N原子与相邻α亚基的Asp221的O1原子形成氢键,Asp221的O2原子与His224的N原子形成氢键,其侧链与单核铁配位。因此,Asp221通过氢键连接两个中心,起到了桥梁作用。如果Asp被其他氨基酸取代,酶会失去所有活性[17]。Pseudomonassp.NCIB 9816-4萘双加氧酶的β亚基由6条长且扭曲的混合β片组成,其中3条链的长度为14个氨基酸残基。在折叠层的凹面上有3个螺旋,在折叠层的凸面上有一个附加的N端螺旋,β亚基上最接近铁活性位点和Rieske中心的原子距离大约为10 Å[29]。
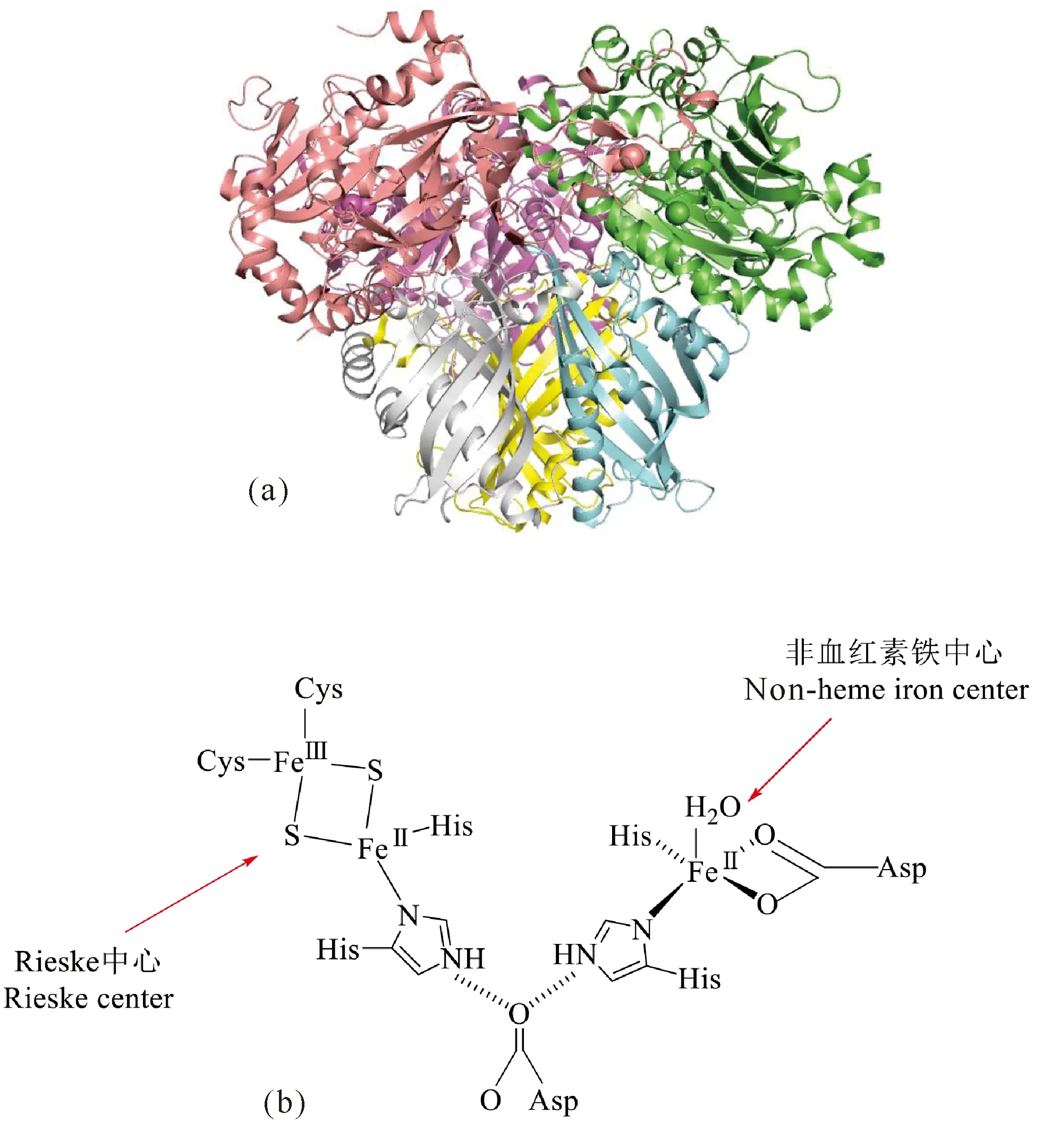
图2 联苯双加氧酶结构及活性中心Fig.2 Structure of biphenyl dioxygenase and its active sites
4 催化机制
目前对萘双加氧酶的催化机制研究较多,萘双加氧酶催化萘降解的初始反应,利用分子氧和NAD(P)H实现萘的顺式1,2-双羟基化[14,37](图3)。黄素蛋白还原酶与铁氧还原蛋白共同组成电子传递链,将来自NAD(P)H的电子转移到末端氧化酶的Rieske[2Fe-2S]中心,铁硫中心接收电子,并将电子传递给单核铁活性位点,该单核铁激活分子氧以插入底物的苯环当中[19]。在没有铁氧还原蛋白的双加氧酶系统中,黄素蛋白还原酶自身存在着与铁氧还原蛋白相似的[2Fe-2S]中心,也能够起到电子传递的作用[38]。铁氧还原蛋白分子质量小,方便移动,大大提高了电子的传递效率,因此双加氧酶系统的演化方向更倾向于铁氧还原蛋白的存在。

图3 萘双加氧酶催化途径[14]Fig.3 Catalytic pathway for naphthalene dioxygenase[14]
在末端氧化酶相邻的α亚基中,保守的Asp作为连接Rieske[2Fe-2S]中心和单核铁中心的桥梁,在催化过程中能够引导电子的转移[39,40]。单核铁中心在静止状态下是一个扭曲的八面体几何结构[41],底物与酶活性位点的结合导致亚铁离子配位几何结构发生变化。Rieske[2Fe-2S]中心的还原导致一个电子减少,使得连接单核铁中心的天冬氨酸向组氨酸残基移动,并改变了单核铁中心的配位几何结构,为双氧结合提供了足够的空间[42]。联苯双加氧酶晶体结构的研究表明,末端氧化酶在与底物结合时其活性位点的氨基酸残基发生了移动,从而导致空间构象出现了显著的变化[27]。未与底物结合前,活性位点的催化口袋太小,无法容纳联苯分子,且没有进入催化口袋的入口;与底物结合后构象发生变化,从而产生了通向催化口袋的通道,并扩大了催化口袋的空间以容纳联苯。另外,只有在结合了底物且[2Fe-2S]中心被还原后,氧分子才与活性位亚铁离子结合,这种机制可以防止在没有底物的情况下双氧与活性位点的结合,从而保护酶的氧化活性[40]。酶蛋白晶体结构的解析能够更深层次地揭示催化口袋在底物结合时发生的重要构象变化,探讨底物进入活性中心的模式。
5 蛋白质工程
蛋白质工程已经被用来提高Rieske型芳香环双加氧酶的催化速率、底物特异性和立体选择性。依据目前对酶结构和功能的认识,设计改造双加氧酶是优化和提升微生物生物催化能力的有效手段。基于活性位点的定点诱变和基因重组技术可以获得更加高效降解芳香族化合物的双加氧酶突变体(表3)。Barriault等[30]已经证明联苯双加氧酶α亚基中氨基酸与底物识别有关,定点诱变α亚基中氨基酸,发现部分氨基酸的取代能够改变酶对底物的特异性或提高酶的催化效率。这些氨基酸的位置靠近单核铁活性中心,可能在酶的催化活性中起一定的作用。对Burkholderia2,4-二硝基甲苯双加氧酶(DDO)α亚基的I204位点饱和突变,突变体DDO提高了对2,4-和2,6-对二硝基甲苯的氧化能力[43]。Bernath-Levin等[44]通过多种蛋白质工程技术得到硝基苯双加氧酶突变体,其催化能力是野生型的3倍。突变体主要通过增大底物结合口袋空间和改变底物结合模式的方式提高酶的活性。对Pseudomonassp.NCIB 9816-4萘双加氧酶的诱变研究揭示,活性位点的几何形状变化与酶的结构-功能存在关系,在F202,A206,V260,H295,F352和L307位点引入单点突变会导致酶的底物特异性、区域选择性和立体选择性发生剧烈变化,但同时保留对天然底物萘的活性[13]。同样地,通过改变一个活性位点氨基酸侧链的大小,能够改变酶的区域选择性和立体选择性[12]。此外,利用基因重组技术可以将一个双加氧酶α亚基碳端的部分片断替换到另一个双加氧酶α亚基上,获得的新的活性双加氧酶同时具有两个亲本的催化特性[45]。DNA改组技术发现萘双加氧酶的225和407位点残基,对酶的底物特异性产生影响,并可以提高酶对二硝基甲苯的活性[46],另外对联苯双加氧酶DNA的改组实验获得了高效降解多氯联苯及其同系物的基因[47]。

表3 芳香环双加氧酶的蛋白质工程改造Table 3 Protein engineering of aromatic ring dioxygenases
6 应用
芳香族化合物在自然环境中广泛存在,且对生物安全存在一定的危害,如何从环境中有效地消除这些化合物备受关注。微生物利用体内Rieske型芳香环双加氧酶降解芳香族有机污染物是环境微生物修复的重要过程[49]。Rieske型芳香环双加氧酶具有宽泛的底物特异性,能催化多种底物的立体选择性反应,这些特性使它们成为化学工业和医药领域重要手性化合物的合成催化剂,也为研发高效污染物微生物修复技术提供关键基础信息[50]。Narancic等[51]在新分离出的PseudomonasTN301中发现,邻苯二酚在多种邻苯二酚双加氧酶的作用下经连续分解,最后进入三羧酸循环转化为二氧化碳和水。刘如洋[52]将芳香环羟化双加氧酶的编码基因通过基因重组的方式整合到SWH-2菌株上,能够提高该菌对菲的降解能力,同时可将该菌应用于石油污染土壤的生物修复。高纯度手性化合物作为药物中间体在医药领域需求旺盛,而传统化学合成法不具备酶法立体选择性特征,较难获得手性纯化合物,生产成本昂贵[53]。双加氧酶羟基化反应主要产生顺式二氢二醇中间体,该中间体可用于合成手性化合物。Janna等[54]利用Comamonassp.硝基苯双加氧酶突变体能够氧化硫代苯甲醚、对甲苯异氰酸酯等硫化物的能力,合成手性亚砜。尽管这些酶是非常有前途的生物催化剂,但是它们对NAD(P)H的依赖性、多组分性质以及在非细胞系统中的固有不稳定性阻碍其实际应用,目前大多数利用Rieske型芳香环双加氧酶合成化学品的应用主要依靠全细胞催化[55]。
7 展望
Rieske型芳香环双加氧酶来源丰富,是一类广泛存在的多组分酶,其介导的催化反应不仅是有机污染物降解的重要途径,也可用于生产制备重要手性化合物中间体。目前关于酶的组分、晶体机构和催化原理已有所了解,但是仍有许多细节值得深入研究。例如酶组分之间的相互作用、电子传递过程中活性位点的铁离子络合物的变化,以及不同氨基酸残基与底物如何相互作用从而影响酶的特异性等。
基因组学的飞速发展使得更多的双加氧酶家族成员被发现,极大地扩展了该类型酶的底物谱与催化反应范围,为更多的化学反应进程研究提供了丰富资源。目前基于计算模拟和分子对接等技术,双加氧酶的催化特性与底物范围研究取得一定的进展,但是在氧化过程中所产生的反应中间体的性质仍有待明确和充分表征。Sutherlin等[56]研究发现,Rieske型芳香环双加氧酶催化反应中存在一种高自旋的FeⅢ过氧化物中间体,而定义这种中间产物的结构对于理解该类酶的化学反应是至关重要的。
未来有关Rieske型芳香环双加氧酶的研究工作不再局限于芳香烃有机污染物的降解,而是基于催化反应的基础研究转向重要手性中间产物的合成。但是该酶系统的多组分复杂性、酶的不稳定性、[2Fe-2S]中心的氧敏感性以及对昂贵的NAD(P)H的共底物需求都给酶促反应带来了巨大的挑战。然而Rieske型芳香环双加氧酶在环境和化学工业领域的巨大应用潜力依然不容忽视,通过蛋白质工程技术优化和改造酶的功能特性,提高其生物催化活性,开发出适用于多种氧化反应的、高效、环保、廉价的生物催化剂,仍是未来发展的趋势。
猜你喜欢
中老年保健(2022年3期)2022-08-24
化工设计通讯(2021年4期)2021-01-07
世界最新医学信息文摘(2020年68期)2020-12-25
中国材料进展(2019年10期)2019-12-07
中国食用菌(2019年7期)2019-08-13
食用菌(2019年2期)2019-04-23
延安大学学报(医学科学版)(2019年1期)2019-03-29
中国药理学与毒理学杂志(2015年3期)2015-12-16
郑州大学学报(理学版)(2014年2期)2014-03-01
现代检验医学杂志(2014年1期)2014-02-06
