
加米霉素的合成及其晶体结构测定
2020-11-18 10:20:58崔志刚王建甄盼盼余贵菊杨雪王猛姜淋洁娄艳华程雪娇
中国兽药杂志 2020年9期
崔志刚,王建,甄盼盼,余贵菊,杨雪,王猛,姜淋洁,娄艳华,程雪娇
(天津市中升挑战生物科技有限公司, 天津 300380)
加米霉素[1](Gamithromycin) 化学名(IUPAC):13-[(2,6-二脱氧-3-C-甲基-3-O-甲基-α-L-吡喃核糖基) 氧 ]-2-乙基-3,4,10-三脱氧3,5,8,10,12,14-六甲基-7-丙基-11-[[3,4,6-三脱氧-3-(二甲氨基)-β-D-吡喃木糖基]氧]-(2R,3S,4R,5S,8R,10R,11R,12S,13S,14R)-1-氧杂-7-氮杂环-15-单环。分子式:C40H76N2O12,CAS号:145435-72-9,是一种新型的第二代大环内酯类兽用抗生素。法国梅里亚公司(MERIAL.CO)生产的以加米霉素为主要有效成分的注射剂ZACTRAN,经欧盟药品监督管理局兽药署的审核,已经实现市场化生产和销售,加米霉素结构和功效已经得到了欧盟药监局兽药署的认可,可用于食用动物的食用,具有吸收快、体内分布广泛、体内残留低、安全性高等优点,暂无不良反应报道,是一种明确可以使用的兽用抗生素,在兽医临床具有广阔的应用前景,目前国内很多厂家已成功申报二类新兽药。对于作为药物或保健食品成分使用的化合物而言,晶体结构对化合物的稳定性,生物利用度等指标有重要影响。目前,研究晶体结构最常见和比较准确的方法是X-射线单晶衍射法。文献中已有报道该化合物的合成[2],通常以9-脱氧-9-同型红霉素 A (E)肟为原料,经过构型转化反应,贝克曼重排反应,还原反应,还原胺化反应得到加米霉素,但是仍存在工艺不适合放大生产、不利于环保、生产成本高等问题。基于以上问题,对加米霉素的合成工艺进行优化改进,改变中间体的纯化方法,更换一些反应试剂,从而获得一种更适合放大生产的工艺,并且使整个反应过程更加安全环保,成本更加低廉。同时,首次获得加米霉素单晶,通过X - 射线衍射单晶结构测定其结构,并确定其构型。
1 材 料
1.1 仪器与试剂 XT-4A显微熔点仪,上海济成分析仪器有限公司;ARX 400 MHz核磁共振仪,瑞士Bruker公司;Bruker Smart Apex 单晶衍射仪,德国Bruker AXS 公司。
1.2 主要药品及试剂 氢氧化锂(纯度:98%),无水乙醇(AR),二氯甲烷(AR),丙酮(AR),对甲苯磺酰氯(AR),碳酸氢钠(AR),硼氢化钠(AR),正丙醛(AR,97%),钯碳(含钯5%)(纯度:95%),甲醇(AR),甲基叔丁醚(AR),以上试剂均购于国药集团(天津)有限公司;9 - 脱氧 - 9 - 同型红霉素A(E)肟,批号:130603,纯度:95%,购于浙江国邦药业有限公司;TLC展开剂:二氯甲烷:无水甲醇:氨水 = 20∶1∶0.1。
2 方法与结果
2.1 加米霉素合成路线[3-4]
2.1.1 9-脱氧-9-同型红霉素A(Z)肟的制备[5-7]向500 mL三口瓶中加入9-脱氧-9-同型红霉素 A (E) 肟50.0 g、氢氧化锂4.8 g和无水乙醇500 mL,混合搅拌,室温反应4 h;向反应体系中滴加稀盐酸调节pH至8 ~ 9,然后加水100 mL,析出固体,过滤,烘干;所得固体用150 mL二氯甲烷打浆1 h,过滤,烘干得产品43.5 g,收率: 87%;熔点: 158~162 ℃;1HNMR(CDCl3):5.01(dd,H-13),4.87(d,H-1″),4.40(d,H-1′),3.98(m,H-5″),3.80(s,H-11),3.49(m,H-5),3.27(s,OCH3),3.21(dd,H-2′),2.99(t,H-4″),2.87(m, H-2,H-8,H-10),2.74(m,H-10),2.43(m,H-3′),2.32(d,H-2″),2.27(s,N(CH3)2),1.91(m,H-4),1.87(m,H-14),1.63(m,H-4′),1.37(m6-CH3),1.28(d,10-CH3),1.24(d,5″-CH3),1.18(d-5′-CH3),1.12(d,2-CH3),1.11(s,12-CH3),1.08(d,8-CH3),1.04(d,4-CH3),0.79(t,CH2CH3)。
2.1.2 9-去氧-12-脱氧-9,12-环氧-8a,9-二脱氢-8a-氮杂-8a-高红霉素A的制备 向500 mL三口瓶中加入9-脱氧-9-同型红霉素A(Z)肟40.0 g和200 mL丙酮,搅拌,溶解,降温至-5~5 ℃,滴加含对甲苯磺酰氯20.4 g的丙酮溶液100 mL,搅拌,反应0.5 h,然后滴加三乙胺10 mL,搅拌,反应1.5 h,TLC检测反应完成,过滤,滤饼用200 mL二氯甲烷溶解,无水硫酸钠干燥,过滤,浓缩得到产品37.2 g,收率95.4%,熔点:105~110 ℃;1HNMR(CDCl3):5.17(dd,H-13),4.73(d,H-1″),4.47(d,H-1′),4.15(dq,H-5″),4.09(dd,H-3),3.99(brs,H-5),3.81(t,H-11),3.68(m,H-8),3.65(m,H-5′),3.40(ddd,H-2′),3.23(s,OCH3),2.96(t,H-4″),2.70(m,H-2,H-8,H-10),2.70(OH-10),2.68(m,H-3′),2.57(brd,11-OH),2.45(p,H-2),2.31(s,N(CH3)2),2.28(d,H-2″),2.20(d,4″-OH),2.07(ddq,H-14a),1.90(brd,H-7a),1.75(dd,H-7b),1.74(m,H-4),1.70(m,H-4′),1.69(m,H-14b),1.46(dd,H-2′′),1.40(S,6-CH3),1.29(m,H-4′),1.27(d.10-CH3),1.27(d,5″-CH3),1.25(d,2-CH3),1.24(d,5′-CH3),1.21(S,3″-CH3),1.18(s,12-CH3),1.07(d,8-CH3),1.01(d,4-CH3),0.88(t,CH2CH3)。
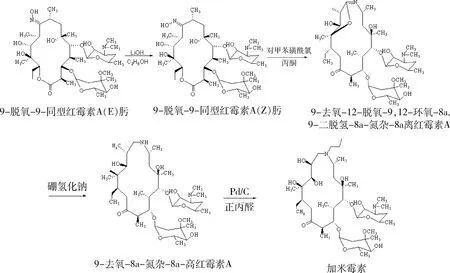
图1 加米霉素合成路线
2.1.3 9-去氧-8a-氮杂-8a-高红霉素A的制备[8-9]
向500 mL三口瓶中加入9-去氧-12-脱氧-9,12-环氧-8a-氮杂-8a-高红霉素A 35.0 g,甲醇175 mL,搅拌溶解,降温-5~5 ℃,分批加入硼氢化钠9.0 g,加毕,搅拌反应2 h,滴加稀盐酸,调节pH至6~7,搅拌反应0.5 h,过滤,浓缩得到粗品9-去氧-8a-氮杂-8a-高红霉素A;将粗品加入到280 mL甲基叔丁醚中,滴加三氟乙酸6.6 g成盐,搅拌1 h,过滤,滤饼加入到200 mL的10%的碳酸钾溶液中,搅拌,加入175 mL二氯甲烷,萃取,无水硫酸钠干燥,浓缩,得到纯品32.9 g;收率:93.5%;熔点:176~180 ℃;1HNMR(CDCl3)5.00(d,H-1″),4.75(dd,H-13),4.48(br,d,H-3),4.34(d,H-1′),4.02(dq,H-5″),3.56(br,H-11),3.52(m,H-5),3.31(s,OCH3),3.16(dd,H-2′),3.01(brd,H-4″),2.78(m,H-8),2.69(dq,H-2),2.59(dd,H-9a),2.42(brt,H-9b),2.30(d,H-2″),2.26(s,N(CH3)2),1.91(m,H-14a),1.77(brp,H-4),1.61(brd,H-4′),1.55(dd,H-2′),1.44(m,H-14b),1.38(m,H-7),1.36(s.6-CH3),1.21(s,3″-CH3),1.20(d,5′-CH3),1.18(d,2-CH3),1.06(s,12-CH3),1.10(d,8-CH3),1.04(d,4-CH3),0.94(d,10-CH3),0.86(d,CH2CH3)。
2.1.4 加米霉素的合成[10]向500 mL高压釜中分别加入9-去氧-8a-氮杂-8a-高红霉素A25 g,正丙醛100 g,冰醋酸3.0 g,搅拌至溶解,加入钯/碳催化剂8.0 g,密封高压釜,釜内用氮气置换三次,氢气置换三次,充入氢气至压力1.4~1.6 MPa,35~45 ℃保温反应,观测压力不再下降。卸压,取样点板,检测原料反应完全,开启釜盖,加入纯化水200 mL,过滤,滤液加入150 mL二氯甲烷,降温至20 ℃以下,用20%盐酸调节pH≤4,萃取分液;水相再加入二氯甲烷150 mL,用20%氢氧化钠溶液调节pH≥10,萃取分液,无水硫酸钠干燥,浓缩,得到加米霉素成品20.0 g,收率:80%;1HNMR(CDCl3):5.00(d,H-1″),4.75(dd,H-13),4.48(br,d,H-3),4.34(d,H-1’),4.02(dq,H-5″),3.56(br,H-11),3.5(m,H-5),3.31(s,OCH3),3.16(dd,H-2′),3.01(brd,H-4″),2.78(m,H-8),2.69(dq,H-2),2.59(dd,H-9a),2.42(brt,H-9b),2.30(d,H-2″),2.26(s,N(CH3)2),1.91(m,H-14a),1.77(brp,H-4),1.61(brd,H-4′),1.55(dd,H-2′),1.44(m,H-14b),1.38(m,H-7),1.36(s.6-CH3),1.21(s,3″-CH3),1.20(d,5′-CH3),1.18(d,2-CH3),1.06(s,12-CH3),1.10(d,8-CH3),1.04(d,4-CH3),0.94(d,10-CH3),0.86(d,CH2CH3)。
2.2 加米霉素的单晶培养及晶体结构测定[11]将1.0 g纯度99%的加米霉素白色固体加入到20 mL乙腈中,70 ℃加热溶解,冷却,静止放置,析出晶体。选取大小为0.22 mm×0.18 mm×0.18 mm的单晶,用Bruker Smart ApexII CCD衍生仪收集衍生数据。
2.2.1 晶体结构描述[12]晶体属于单斜晶系,C2空间群,具有手性,纯度大于99%,晶胞参数如表1和图2所示。

表1 加米霉素晶体结构数据
aR1= Σ || Fo|-| Fc||/| Fo|.bwR2= [ Σw(Fo2-Fc2)2/Σw(Fo2)2]1/2
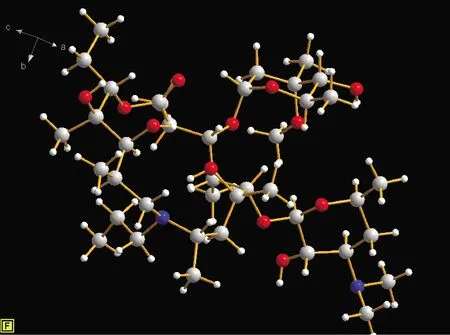
图2 加米霉素的分子立体构型
3 讨 论
本研究以9-脱氧-9-同型红霉素 A (E) 肟为原料,通过构型转化反应,贝克曼重排反应,硼氢化钠还原反应和还原胺化反应得到加米霉素。其中,在构型转化这步反应中,由于在纯化过程中也有可能发生构型转化,因此,寻找一个合适的纯化试剂很重要。本文中采用二氯甲烷进行纯化,原因是原料9-脱氧-9-同型红霉素 A (E) 肟易溶于二氯甲烷, 而9-脱氧-9-同型红霉素 A (Z) 肟不溶于二氯甲烷,通过打浆搅拌的方法使成品纯度在97%以上,高于已报道文献[13]中纯度;该过程操作简单,容易得到纯品,适合大规模生产;文献[13]中,后处理方法采用先萃取再浓缩的方法得到产品,但是在后处理的过程中容易构型再次转变成原料,导致纯度不高。
在制备9-去氧-12-脱氧-9,12-环氧-8a,9-二脱氢-8a-氮杂-8a-高红霉素 A的过程中,如果9-脱氧-9-同型红霉素 A (Z) 肟纯度不高,反应中会产生很多杂质,该步骤也是难度比较大的一个步骤,本文使用三乙胺作为有机碱,不但减少杂质的数量,还减少了对身体的危害,保证了生产的安全性, 该步收率远高于文献[13]报道的收率,文献[13]中所用到的碱为有机碱吡啶及无机碱碳酸氢钠,有机碱吡啶有一定毒性,不适合放大生产,碳酸氢钠会产生一个比较大的杂质,9-羰基-8a-氮杂-8a-同型红霉素 A,很难除去。
在还原反应时,本文后处理未进行纯化操作直接进行还原胺化反应,减少操作步骤,降低产品损失,高于文献收率,工艺经优化后适合放大生产。对比文献[13]采用先还原再纯化的方法,然后再进行胺化反应,导致收率偏低。
加米霉素样品用乙腈作溶剂得到加米霉素单晶,确定了加米霉素晶体的空间点群和构效方式;加米霉素属于大环内酯类化合物,有多个手性中心,会形成很多不同构型,而通过加米霉素单晶解析最终确定了加米霉素的绝对构型,由于该晶体具有很好的晶型结构,对后续研究加米霉素新剂型,丰富临床给药方式,改善药代动力学,减少刺激性提高安全性等方面,具有一定的参考价值。
猜你喜欢
保健与生活(2022年7期)2022-04-08 21:33:36
华声文萃(2021年11期)2021-11-15 18:40:16
中学生数理化(高中版.高考理化)(2020年2期)2020-04-21 05:33:04
国际呼吸杂志(2019年4期)2019-03-12 01:07:30
上海化工(2018年10期)2018-10-31 01:21:06
衡阳师范学院学报(2016年3期)2016-07-10 07:16:27
火炸药学报(2014年3期)2014-03-20 13:17:39
化工生产与技术(2014年5期)2014-02-27 13:42:02
中国应用生理学杂志(2013年2期)2013-03-25 07:59:30
郑州大学学报(理学版)(2013年2期)2013-03-11 20:30:30
