
合成气直接制C2含氧化合物Rh基催化剂中金属-助剂(载体)界面的构建
2020-11-11 06:54陈维苗宋宪根丁云杰
无机化学学报 2020年11期
陈维苗 宋宪根 丁云杰*,,2
(1中国科学院大连化学物理研究所,洁净能源国家实验室(筹),大连 116023)
(2中国科学院大连化学物理研究所,催化重点国家实验室,大连 116023)
0 引 言
随着石油资源的日益匮乏和环保要求的不断提高,由煤、天然气或生物质经合成气制取燃料和化学品具有重大意义[1-6]。由合成气一步法直接制取乙醇、乙醛和乙酸等C2含氧化合物具有工艺流程短、原子经济性高等优点,因而备受关注。该研究在40多年间取得了很大的进展[5-36]。实现一步法合成的关键在于高效催化剂的研发。在已知的催化剂中,Rh基催化剂性能最为优越[9-36],也是最有可能实现工业化的催化剂之一,这源于Rh独特的吸附和活化CO性能[9]。然而,单金属Rh催化剂上CO加氢得到的产物主要是甲烷,因此需要添加助剂和选择合适的载体[5-40],如各类过渡金属(如 Mn[14-21]、V[22-26]、Fe[27-34]、Zr[35]等)、稀土金属(La、Ce、Sm 等)[36-37]以及碱金属或碱土金属[38-39]。尽管Rh基催化剂上CO加氢生成C2含氧化合物的活性位的结构还不是很清楚,且存在很多争议,但有一点是毋庸置疑的,即该活性中心必定是在活性金属与助剂的界面处。因此,Rh与助剂必须紧密接触,以创造出更多的金属-助剂(载体)界面面积[4,19,40],这样才能最大限度地发挥贵金属Rh的催化效率,从而有利于推动该过程的工业化。
1 金属-助剂(载体)界面的性质
当负载的金属颗粒粒径从10 nm降至1 nm时,存在于不同局部环境中的暴露的金属原子比例会发生变化,如暴露的配位不饱和的金属原子比例会增加,其形貌也会发生改变。这也会影响金属-助剂界面处的金属原子,导致其比例增加;电荷转移主要影响界面处金属原子的性质,乃至其周边活性点位的性质[42]。但当金属粒径降至1 nm以下时,金属原有的电子结构会发生很大改变,它们与载体/助剂的相互作用以及界面处金属原子的性质与大粒径时的可能完全不同,因此表现出不同的活性-粒径的对应关系。正是由于界面处的金属和助剂原子所处的配位、几何以及电子环境与别处的不同,造成其界面金属活性位表现出不同的吸附和活化反应物能力,从而导致其具有独特的催化性能[40]。可见,界面处的金属活性位在反应中不仅仅具有双功能催化的作用。根据Rh基催化剂上CO加氢生成C2含氧化合物的机理可知[41],解离活化和非解离活化CO能力必须相互匹配:前者一般发生在Rh-助剂形成的界面上,而后者在孤立的Rh中心上。在反应过程中,非解离CO必须尽快插入到CO解离后的物种CHx中才能生成C2含氧化合物前驱物种,否则CHx会继续链增长,因此,这2种主要活性中心必须足够接近,即二者处在界面处,相互协同,共同催化目标反应的进行。
可见,采用有效的方法构建出浓度尽可能高的活性位,并深入理解和认识其结构和动态性能对研制出高性能的催化剂至关重要。我们重点介绍现有的,特别是最近出现的一些最大化金属-助剂(载体)间界面的原位或体外的方法,通过控制金属-助剂界面的数量和性质来调变催化剂性能,最终找到产生仅存在于金属-助剂界面的均一分布的活性位的方法,并对该领域存在的挑战和研究前沿进行展望。
需要注意的是,当载体为可还原性金属氧化物时,载体因催化剂还原而与活性金属发生相互作用,在二者界面创造出反应活性位,其修饰效果与助剂是类似的。因此我们所指的界面包含这2种情况,但以助剂-金属间界面为主。
2 金属-助剂(载体)界面构建方法
2.1 金属-载体强相互作用(SMSI)涂覆效应
Tauster等[43]首次发现Rh/TiO2催化剂经过高温还原后存在SMSI,其它可被还原的氧化物(如V2O5和La2O3等)用作催化剂载体时同样存在该现象[44-45]。这是由于在高温还原的过程中形成了低价氧化物,它覆盖了部分金属颗粒,降低了金属的有效面积,虽然导致H2和CO的化学吸附量显著减少,但可显著提高CO加氢反应活性和生成C2含氧化合物的选择性。
SMSI效应可推广至可还原的氧化物做助剂的情况,例如V2O5,当它被用作载体时存在SMSI效应;当它被用作Rh/SiO2催化剂的助剂时,在催化剂的焙烧过程中能够形成RhVO4相,并且在随后250℃的还原中会导致Rh表面被V2O3物相所覆盖,如图1B和E所示。当Al2O3被用作载体时,仅仅当V/Rh值很高时,才能看到相似的现象。对于这些催化剂来说,Rh可能位于层状的V2O5上,经过还原后一些钒的氧化物可能迁移到Rh粒子上,如图1C和1F所示。在具有低的V/Rh值的催化剂上,Rh和V2O3被认为是以孤立的颗粒存在于Al2O3载体表面,这是由于钒的氧化物与Al2O3载体之间具有强烈的相互作用[23]。一般认为,CO的解离需要较大的金属颗粒,而插入过程则不需要。因此,当金属表面由于SMSI作用被部分覆盖时,活性金属被稀释,从而抑制了甲烷的生成,提高了含氧化合物的选择性。可见,正是由于SMSI的存在使得助剂甚至于载体与活性金属的接触更多了,对Rh的修饰作用就更明显,从而产生了更多的助剂(或载体)与Rh接触面,形成邻近的CO插入或解离活性位,这大大提高了C2含氧化合物的选择性。研究发现,在利用SMSI形成更多接触面积时,控制好催化剂中助剂与Rh的相对含量以及适宜的还原温度才能达到更好的修饰效果[46]。
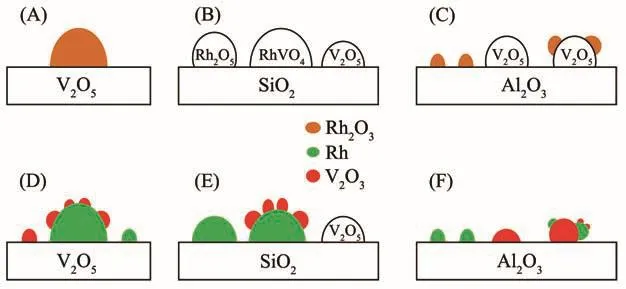
图1 不同类型载体负载的VOx-Rh催化剂经350℃焙烧 (A~C)以及250℃还原 (D~F)后的表面状态[23]Fig.1 Schematic of surface state of VOx-Rh catalyst over different supports after calcination at 350℃(A~C)and reduction by H2 at 250℃ (D~F)[23]
综上所述,通过高温H2还原法可以实现助剂或载体氧化物对金属Rh表面的部分修饰,形成SMSI涂覆层,从而产生高浓度的界面活性位。但研究也发现,这种结构在反应过程中不稳定,因此在研究SMSI效应的构效关系时存在很大问题[46-47]。最近,Matsubu等[48]利用反应物诱导SMSI效应在6%Rh/TiO2(6%为质量分数)催化剂上形成了多孔的涂覆层(在体积分数为20%CO2和2%H2的气氛中,250℃、100 kPa条件下处理)。通过原位透射电镜(TEM)证实,与高温H2还原形成的结构不同,该涂覆层结构可稳定存在于水气氛中。在CO2加氢反应中,该催化剂的CO选择性达90%,而常规法制备的催化剂的催化产物全为甲烷。该结果清楚地证实,因载体或助剂的还原而形成于金属涂覆层中的暴露的活性位点确实是处于界面处,而常规法制备的催化剂的活性位基本在独立的金属或助剂上。这种方法的应用对象只限于具有可还原性的氧化物助剂或载体,因而在实际应用中还存在很大的挑战。另外,通过采用适当的原位表征方法,如原位TEM、电子能量损失谱(EELS)等,并将该表征方法中所采用的高能电子束辐射对样品结构的影响降至最低[49-50],同时结合红外、X射线吸收光谱(XAS)等常规方法以及密度泛函理论(DFT)计算,进一步深入准确研究涂覆层的化学组成、形貌、结构性质和稳定性,准确建立起它们与反应性能之间的对应关系[40,51]。
2.2 形成复合氧化物的形成
Kunimori课题组发现[24-26],在硅胶载体上通过Rh与过渡金属氧化物在O2或空气中高温(700~900℃)焙烧,可得到含Rh、过渡金属和氧的三元化合物,如 RhNbO4、RhVO4和 Rh2MnO4,其对负载型 Rh的形貌和催化性能的影响很大。该研究表明(图2):(1)通过300 ℃以上的H2还原,RhVO4和Rh2MnO4分解后生成的Rh呈高度分散状态;(2)Rh与助剂氧化物间产生强相互作用。Rh与金属氧化物MOx之间紧密接触,界面积达到最优,充分发挥助剂的作用;(3)通过再次焙烧后得到复合氧化物,再经H2的还原,Rh金属重新再分散,其催化性能也得到恢复,该过程可重复8次,且其催化性能保持不变[26]。在用作助剂的过渡金属能与Rh形成复合氧化物且Rh能高度分散于载体上的情况下,该方法才能适用;能在温和的条件下与H2反应,分解成Rh与过渡金属氧化物。而Rh与过渡金属氧化物之间能紧密接触,产生CO解离和插入的活性位。2种活性位的位置足够近使得解离后形成的CHx插入到邻近的CO中。另外,形成的复合氧化物中Rh与过渡金属之间的比例应适当。实际上,能满足这些要求的助剂不多,因此,这种方法存在很大的局限性。
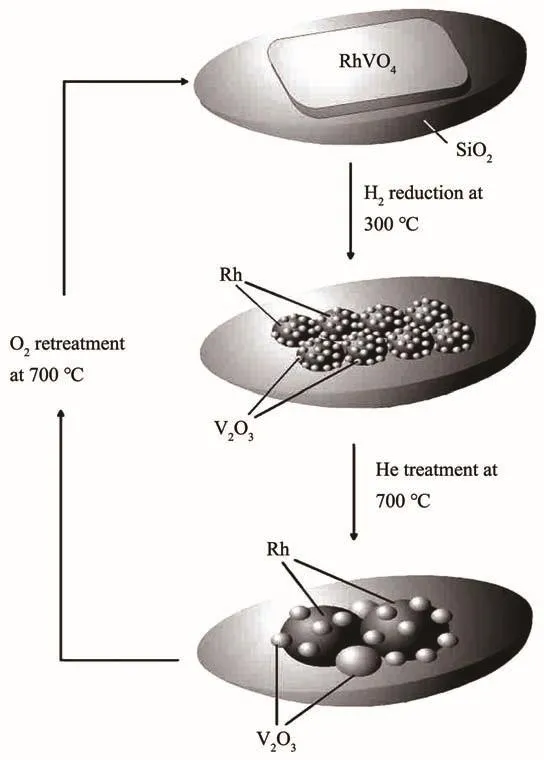
图2 RhVO4的再生与Rh在硅胶载体上的再分散模型[26]Fig.2 Model for regeneration of RhVO4 and redispersion of Rh on SiO2 support[26]
2.3 单层分散
一些氧化物载体,如Al2O3具有较高的活性表面羟基密度,有利于形成负载金属氧化物的单覆盖层,从而优化金属Rh与助剂金属氧化物之间的相互作用。Burch等[27]研究了不同Fe载量的2%Rh-Fe/Al2O3(如无特别说明,后文百分含量均指质量分数)催化剂上CO的加氢反应性能。当Fe载量为10%时,氧化铁在Al2O3表面形成单覆盖层,使其与Rh间的相互作用最佳,Rh-FeOx界面面积达到最大,同时该界面间的CO吸附模式影响了产物选择性(有利于使生成的C2含氧化合物的活性中心数量最多),此时乙醇选择性可达50%以上。进一步研究发现[28],Rh-Fe/Al2O3催化剂表面可能存在Rh-Feδ+-O活性中心和单独Fe物种,前者与乙醇等C2含氧化合物的生成有关,而后者则影响甲醇选择性;另外,载体焙烧温度可有效调节载体与Rh和Fe间相互作用,从而改变了上述活性中心的相对数量,乃至影响所得催化剂上CO加氢生成乙醇的反应性能。这种方法可提高Rh的利用效率,然而,助剂除了与活性金属Rh形成生成C2含氧化合物的活性中心外,更多的是不与Rh接触,形成单独的活性中心,起着加氢或链增长的作用,促进了副产物的生成,从而导致C2含氧化合物选择性下降。
2.4 合金
由于在Rh基催化的助剂中,Fe物种比较容易还原到金属态,因此,目前报道最多的存在于负载型Rh基催化剂中的合金是Rh-Fe合金。在合金结构中,2种组分一定是紧密接触的。Schunemann等[29-30]发现,NaY分子筛负载的2.9%Rh-1.94%Fe催化剂中Fe以Fe0和Fe2+形式存在:其中Fe0可与Rh0形成合金,从而提高催化剂活性,而Fe2+作为一个单独的氧化物相,与Rh颗粒的紧密接触可提高CO加氢生成C2含氧化合物的选择性。由于该催化剂中Rh和Fe含量相对较高,在Fe0的含量较高时其很容易与Rh形成合金。这是首次有关助剂与Rh形成合金有利于提高CO加氢反应性能的报道。
Palomino等[31]详细研究了TiO2负载的Rh-Fe催化剂在还原和反应过程中各物种的演变和Fe-Rh合金的形成规律及其对CO加氢反应性能的影响。结果发现,许多金属Fe在反应过程中经历了碳化和氧化及存在金属Rh的现象说明Fe与Rh形成了表面合金,内核是纯Rh。进一步研究发现,当Fe含量低时,部分Fe进入到Rh纳米粒子中形成表面Fe-Rh合金,剩余的Fe以氧化态的形式存在于Rh颗粒的附近或载体上。随着Fe含量增加到4%,合金的浓度增加;当Fe含量高于4%时则有利于形成金属Fe。在CO加氢反应过程中,金属Fe沉积下来,而合金中的Fe转化为氧化物和碳化物相。结合在反应过程中催化剂各物相浓度与反应性能的关系发现,FeOx在乙醇合成中起着重大的作用。在CO转化率最低时乙醇选择性最高、甲烷选择性最低,这表明Fe-Rh合金和FeOx的增强作用表现为修饰或堵塞Rh位。Rh-Fe合金中Fe与Rh的紧密接触改变了活性Rh位的数目和性质,使得乙醇的生成速率超过甲烷。DFT计算结果表明,决定Fe修饰的Rh表面上乙醇选择性的关键因素是甲基加氢和CO插入的相对能垒,后者是乙醇生成的关键步骤[52]。
上述研究中虽然采用原位X射线衍射(XRD)揭示了FexRh1-x合金相的存在,但由于其衍射峰与载体TiO2重叠,同时在反应过程中还检测到FeOx相,且该物相催化作用也未确定,因此,Carrillo等[32]采用纳米Fe2O3作为载体和Fe源用于原位形成FeRh合金,以完善以上研究的不足之处(图3)。该研究利用Fe2O3的可还原性和高比表面积使Rh-Fe2O3界面最大化,以促进在还原性气氛(H2或合成气)中FeRh合金的生成。原位X射线吸收近边结构(XANES)结果表明,Fe和Rh在催化反应时以金属态存在,但当从还原态变为反应态时,Fe的化学态发生了变化。原位XRD结果表明,在还原条件下,Rh通过形成FeRh合金进入α-Fe的bcc单位晶胞中,合金结构式可表示为Fe1-xRhx(0.06≤x≤0.5),其结构组成取决于Rh-Fe界面的多样性和Rh粒径。在反应条件下,由于Rh的存在,使得载体完全转化为金属Fe和碳化物(Fe2.2C)(图3)。定量计算得出,质量分数为2%Rh的样品中80%Rh与Fe形成合金;随着Rh含量增加,该比例降至60%,这可能是由于还原和反应温度的降低不利于合金的形成。总体而言,随着Rh含量增加,样品中FeRh合金的含量增加(图4)。另外,作者还通过原位扩展X射线吸收谱精细结构(EXAFS)给出了合金存在的证据。Ichikawa等[33]曾利用EXAFS检测了Fe和Rh还原后的Fe-Rh/SiO2催化剂,其中在Fe的EXAFS中测得Fe-Rh键长为0.262 nm,但在RhK边的EXAFS未检测到Rh-Fe键的存在。Gogate等[34]在Fe和Rh的EXAFS均未在反应过程中检测到Fe-Rh/TiO2催化剂中Fe-Rh合金的存在。总之,Carrillo等[32]利用原位XRD和EXAFS数据清楚地证实了在反应过程中金属态FeRh合金的存在。结合反应结果可知,乙醇等含氧化合物选择性的升高与FeRh合金有关,而Fe2O3与碳化铁物相则与烃类和甲烷的形成有关。Huang等[19]采用原子级的表征手段(如球差电镜、X射线能谱分析和EELS谱等)在Rh-Mn-Fe/SiO2催化剂中也检测到了Rh-Fe合金的存在且助剂与金属是紧密接触的。
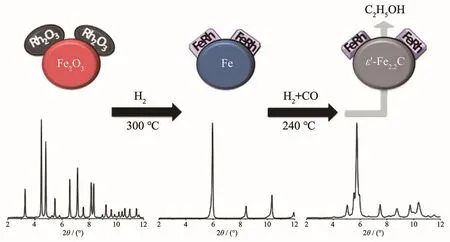
图3 用于含氧化合物合成的FeRh纳米合金的原位形成[32]Fig.3 In situ formation of FeRh nanoalloys for oxygenate synthesis[32]

图4 Rh/nFe2O3催化剂上CO加氢反应生成含氧化合物选择性和其中各类Fe物相含量与Rh含量的关系[32]Fig.4 Oxygenate selectivity(right)and iron phase content extracted from Rietveld refinements(left)versus Rh loading for Rh/nFe2O3 catalysts studied under CO hydrogenation conditions after hydrogen reduction[32]
另外,Rh-Mn催化剂中也可能存在Rh-Mn合金。Mei等[20]结合实验和基于第一性原理的动力学模拟研究了硅胶负载的Rh/Mn合金催化剂上CO加氢生成乙醇反应。X射线光电子能谱(XPS)、TEM和XRD等表征结果都证实了催化剂中形成了Rh/Mn合金,这是C2含氧化合物的生成活性位。理论计算结果也表明,在还原性的反应气氛中,Rh/Mn二元合金的热力学稳定性要高于Rh-MnOx混合氧化物。尽管助剂Mn与Rh形成的二元合金不影响甲烷的生成能垒,CO插入CH2和CH3的反应能垒仍较高,但却降低了CO插入CH反应能垒,从而使得乙醇等C2含氧化合物的选择性增加。理论结果和实验结果吻合较好。但在实际中氧化锰很难还原为金属,因此形成Rh-Mn合金的可能性较小[19]。
总之,相对较易还原的Fe物种促进的Rh基催化剂中容易形成Rh-Fe合金,由于二组分足够接近,产生了CO加氢生成C2含氧化合物的生成活性位,各活性位的功能协同作用,共同催化CO加氢反应的进行。
2.5 强静电吸附(SEA)法
SEA法指在浸渍过程中调节金属离子与带电荷的载体之间的静电相互作用,从而改进助剂与金属的相互作用,以提高界面面积的方法。该方法依赖于当氧化物载体与酸性或碱性的浸渍溶液相接触时其表面所带的电荷:载体表面羟基若质子化则带正电荷,脱质子化则带负电荷。在给定pH值下载体表面带电荷的羟基浓度与其等电点(PZC,表面羟基不带电荷时的pH值)有关。如碱性PZC的氧化物处在酸性溶液中时会产生更高浓度的质子化的羟基(OH2+),这将会吸附阴离子的配合物或金属前驱体,反之亦然。因此,在某一pH值下助剂前驱体会优先吸附在活性金属氧化物表面,而不是载体上,从而可以产生更多金属-助剂接触面。Liu等[21]利用MnO4-离子在酸性的pH值下选择性地吸附在Rh2O3上(PZC=8.75),而不是吸附在具有低PZC的硅胶(PZC=4)上,从而制得Mn促进的Rh/SiO2催化剂(1%MnSEA3%Rh/SiO2),如图5所示。扫描透射电镜(STEM)-EELS谱揭示,与常规浸渍法制得的1%MnIWI3%Rh/SiO2催化剂相比,该催化剂中Rh-Mn间相互作用程度更高;XANES谱和程序升温还原结果表明,助剂Mn的加入对该催化剂中Rh性质的影响最小。在CO加氢反应中,SEA催化剂表现出高的乙醇选择性(24%vs 6.4%),如图6所示。这表明助剂与金属间紧密的相互接触是提高乙醇选择性的关键因素,生成的乙醇活性位位于金属-助剂的界面,该界面越大,其生成效率越高。
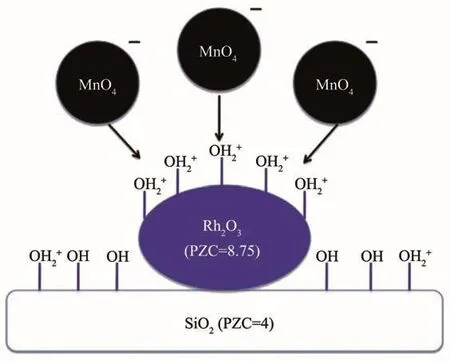
图5 酸性溶液中Mn助剂选择性吸附到Rh催化剂上的示意图[21]Fig.5 Schematic of the selective adsorption of Mn promoters onto supported rhodium catalysts in acidic solution[21]
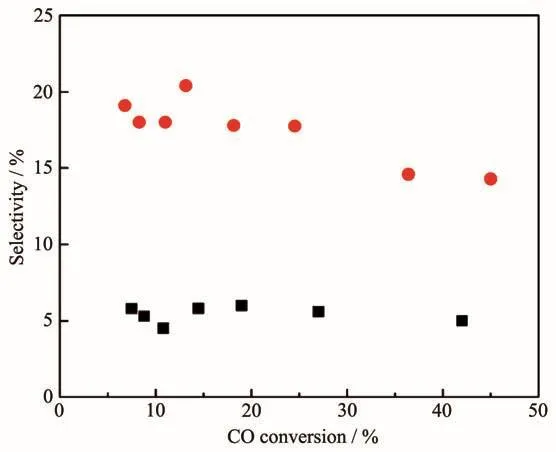
图6 1%MnSEA3%Rh/SiO2和1%MnIWI3%Rh/SiO2催化剂上乙醇选择性与CO转化率的关系[21]Fig.6 Ethanol selectivities at various CO conversion points for the 1%MnSEA3%Rh/SiO2 catalyst and 1%MnIWI3%Rh/SiO2 catalyst[21]
Ro等[53]在适当的浸渍液pH值(高于ReOx的PZC但低于氧化铝的PZC)下,采用SEA法选择性地将Rh前驱物种[(H2NCH2CH2NH2)3Rh]3+沉积到ReOx上而不是载体氧化铝上,从而促进了Rh与Re之间的接触作用,得到原子分散的多原子物种Rh-ReOx(图7),该催化剂在乙烯氢甲酰化反应中表现出高的性能。
SEA法需要适宜的调节剂以精确控制浸渍液的pH值,同时还要求Rh配合物具有多变的价态和适宜的正负电荷,这就意味着需要选择复杂的螯合剂,而这些条件在实际应用过程中很难达到。
2.6 控制表面反应(CSR)法
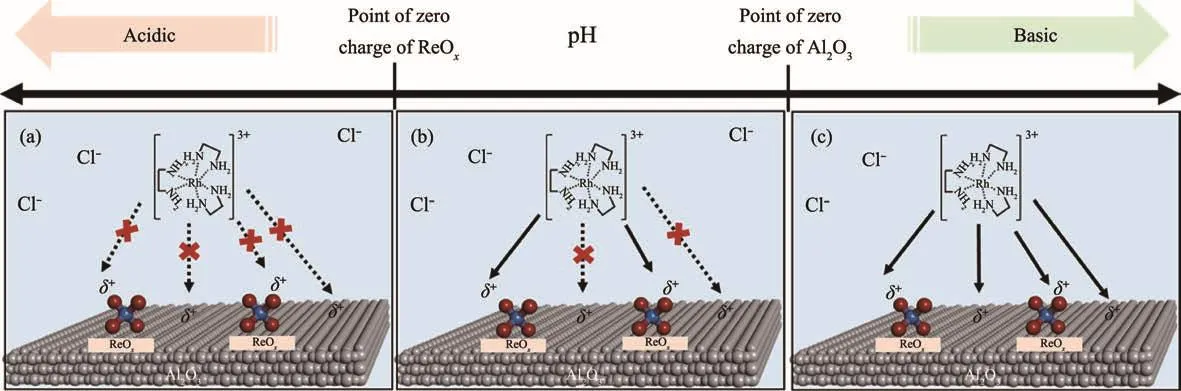
图7 用SEA法将Rh前驱体有选择性地沉积到ReOx附近区域的示意图[44]Fig.7 Schematic of the SEA-based synthetic protocol allowing selective deposition of the cationic Rh precursor([(H2NCH2CH2NH2)3Rh]3+)near ReOxdomains via electrostatic attraction between metal oxide surfaces and charged precursors in solution at three different pH regions[44]
CSR法可通过将助剂选择性地沉积到活性金属表面而不是在载体上,样品具有较窄的粒径分布和可控的组成,从而制得助剂-金属紧密接触的催化剂[16,54],是一种制备高浓度的结构确定的界面活性位的方法。Liu等[16]采用CSR法将Fe和Mn物种选择性地锚合在硅胶负载的Rh纳米颗粒上。先通过浸渍、干燥和还原得到Rh/SiO2催化剂,降至室温后密封于惰性气体保护的Schlenk管中,再转移至手套箱中,将环己二烯三羰基铁或环戊二烯三羰基锰的正戊烷溶液加入管中,搅拌2 h后除去溶液,利用紫外-可见光(UV-Vis)光谱测得Fe和Mn前驱体的吸附量。UV-Vis吸收光谱、STEM和电感耦合等离子体发射光谱结果表明,Fe和Mn前驱体没有与载体发生作用,且2种前驱体之间的作用也很小,而它们与Rh的相互作用、可利用的Rh位以及Fe与Mn的沉积次序影响了Mn和Fe的沉积量。Rh与Fe的紧密接触促进了乙醇等C2含氧化合物的生成,而Rh与Mn的相互作用则提高了整体反应活性和C2+烃类选择性。Mn与Fe同时与Rh紧密接触,使催化剂活性和选择性进一步提高。XPS结果表明,催化剂还原后相当多的Fe和Mn物种分别以金属Fe和MnOx的形式存在于金属Rh表面。DFT结果证实,Mn以Mn(Ⅰ)和Mn(Ⅱ)氧化物形式存在于Rh(211)面台阶边缘;Fe以Fe(Ⅰ)氧化物形式位于Rh(211)面台阶边缘,而金属次表层Fe位于Rh(211)面的台阶和平台处。遗憾的是,作者并未比较常规浸渍法制得的催化剂的性能,但我们对Mn和Fe助催化作用的讨论则因它们与载体无相互作用而显得更加纯粹。Aragao等[55]采用CSR法制备Pt-Fe/SiO2催化剂时发现,Fe前驱体螯合物的几何形状和不饱和度影响了Fe在金属Pt上的选择性沉积,一旦沉积发生,将因氧化而形成Pt-FeOx界面位,与下文讨论的原子层沉积(ALD)法选择性沉积制备的催化剂很类似。
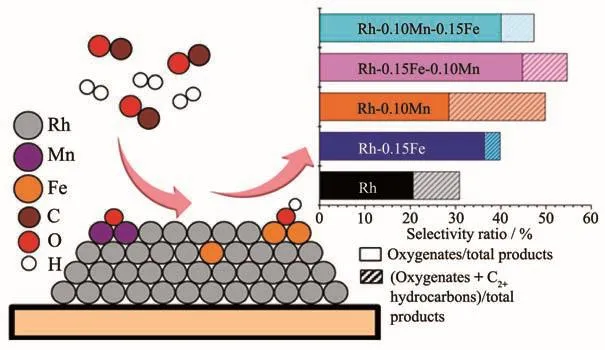
图8 CSR法制备的Fe和Mn助催化的催化剂上的CO加氢反应[16]Fig.8 Synthesis gas conversion over Fe and Mn promoted Rh/SiO2 catalysts prepared by CSR method[16]
由于该方法需要设计和筛选结构较为复杂的金属螯合物,使得实际操作变得很不方便。
2.7 ALD法
ALD法可将物质以单原子膜形式一层一层地镀在基底表面。它利用限制性表面反应在原子精度上控制形成均匀的沉积材料,在诸多领域均有应用[56]。最初在催化领域采用该技术是为了在金属纳米颗粒表面形成多孔的氧化物膜,通过氧化物膜抑制金属移动,防止金属因积碳和烧结而失活,从而提高催化剂热稳定性[57-58]。后来人们发现这些涂层还可通过堵塞活性位而非产生金属-载体界面来提高催化剂的活性和选择性[59]。由于这种技术涂覆的氧化物层不仅涂覆在金属上,而且也沉积在载体上。因此,采用ALD技术选择性地将氧化物层沉积到金属上而不是同时沉积在金属和载体上显得意义更大。最近,Singh等[60]改进了该技术,采用O2作为共反应物,可以有选择性地将金属氧化物沉积在金属纳米粒子上。
Yang等[17]在浸渍Rh之前,采用ALD法将该MnO沉积到载体硅胶上,得到Rh/MnO/SiO2催化剂。为了比较,再采用ALD法将超薄的MnO层选择性沉积到Rh/SiO2上,制得MnO/Rh/SiO2催化剂。同时采用共浸渍法制得Rh-Mn/SiO2催化剂。结果表明,在不改变Rh/MnO/SiO2催化剂载体结构的情况下,预先沉积的MnO薄层并不影响Rh颗粒的粒径和还原性能。因此,催化剂性能的变化主要来自于Rh-MnO的界面位,在此形成倾斜式CO吸附物种,促进了CO解离,因而活性更高;同时,Rh-MnO界面的存在稳定了CO加氢反应过程形成的乙酰基中间体,从而提高了C2含氧化合物的选择性。实验还发现,Rh与MnO之间大的接触面积更有利于提高催化剂选择性和单位数量Rh的活性。对于MnO/Rh/SiO2催化剂,由于在吸附CO时,沉积在金属Rh上的MnO因不稳定而发生了迁移,这与SMSI效应很类似,即涂覆在金属上的部分还原的载体氧化物在反应条件下也不稳定,易从金属上迁移下来。共浸渍法制得的催化剂性能也较高,但其中Mn的作用不仅仅是形成了界面,还有其他的效应,如有助于形成更小的Rh粒径、更低的Rh还原性,由于Mn和Rh混合得很好,也不能排除形成复合氧化物和合金的可能性。
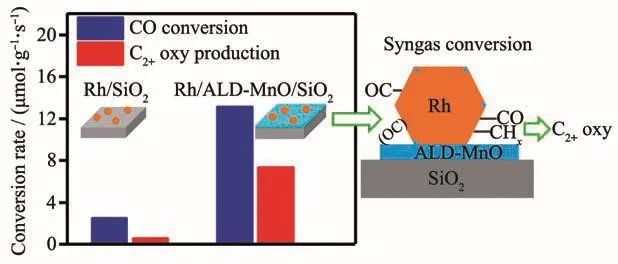
图9 ALD法构建的Rh-MnO界面位促进合成气转化为含氧化合物[18]Fig.9 Rh-MnO interface sites formed by ALD method promote syngas conversion to higher oxygenates[18]
Liu等[18]采用ALD法在硅胶载体上涂覆WxC,可有效抑制主要副产物甲烷的生成,从而提高Rh-Mn/SiO2催化剂上CO加氢生成C2含氧化合物的性能。Zhang等[61]也尝试用ALD法在硅胶涂覆MoO3、α-MoC、β-Mo2C、W2C或碳化钼,然后再浸渍Rh制得催化剂。结果表明,涂覆层可有效抑制甲烷的生成,促进甲醇的生成,提高CO的转化率,另外涂覆层可有效消除载体中杂质对催化性能的影响。其中Rh/MoO3/SiO2催化剂性能最好,甲烷选择性为11%,含氧物选择性为84%。表征结果发现,金属Rh吸附的CO物种主要为(211)面上的桥式,这有利于甲烷的生成;当涂覆MoO3后CO吸附物种主要为孪式和少量的桥式,这有利于甲醇的生成。
3 结论与展望
随着含有原子尺度金属物种和具有性质可调的多孔氧化物覆盖层的催化剂合成技术的发展,通过上述合成方法或各方法相互之间的融合,使得助剂(或载体)有选择性地、最大化地与活性金属相互作用,很可能制备出均一分布、界面活性位结构确定的催化剂。由于CO加氢生成C2含氧化合物的反应发生在Rh-助剂界面,因此最大化其界面处的接触面积一直是人们研发高性能Rh基催化剂的重要突破口。最近人们正在不断尝试新的合成方法来提高Rh-助剂或载体界面积。虽然我们所论述的这些方法合成的催化剂性能与常规浸渍催化剂相比并无明显优势,且只在特定领域才会有应用,在实际催化剂的生产中仍具有很大的挑战性,但此类研究却迈出了坚实的探索性步伐,同时在解释助剂作用时显得更有说服力,因为这类催化剂排除了其他因素的干扰,有助于更深地理解界面处的结构及物化性质,从而帮助我们开发出性能更高、成本更低的Rh基催化剂;同时随着各类技术的不断进步,有些方法,如ALD法在该领域将会发挥很大的作用。另外,由于在反应过程中,金属、助剂或载体的结构性质是动态的,其界面处更是如此,因此要想更深入了解界面处性质与催化性能的构效关系,需要采用超快的检测表征方法。
猜你喜欢
纺织标准与质量(2022年3期)2022-08-10
中学化学(2022年5期)2022-06-17
陶瓷学报(2021年4期)2021-10-14
陶瓷学报(2021年1期)2021-04-13
陶瓷学报(2021年1期)2021-04-13
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
今日农业(2020年20期)2020-11-26
中学生数理化·高二版(2016年3期)2016-12-26
中学生数理化·高二版(2016年3期)2016-12-26
山东工业技术(2016年15期)2016-12-01
