
稳定表达Cas9 蛋白的HeLa 细胞系的建立
2020-11-10 00:44刘燕飞王晓钧
中国预防兽医学报 2020年9期
刘燕飞,那 雷,王晓钧
(中国农业科学院哈尔滨兽医研究所兽医生物技术国家重点实验室/马传染病和慢病毒病研究创新团队,黑龙江 哈尔滨 150069)
CRISPR(Clustered regularly interspersed short pal⁃indromic repeat)是大肠杆菌基因组存在的规律成簇的间隔短回文序列[1-2]。在CRISPR序列位点附近存在一类具有多态性的家族基因,其编码的蛋白具有核酸酶活性,可与CRISPR 序列共同抵御外界噬菌体和外源基因的侵入。这些蛋白称为CRISPR 相关蛋白(CRISPR associated,Cas),与CRISPR 序列组成的CRISPR-Cas 系统是细菌中重要的获得性免疫系统[3]。
目前CRISPR-Cas 系统分为3 种类型,Ⅰ型、Ⅱ型和Ⅲ型。CRISPR-Cas9 系统属于Ⅱ型,与其它两种类型相比,该系统只需要一个蛋白即Cas9 蛋白参与便可发挥其免疫功能,因此其机制研究得最为清楚,应用也最为广泛。当外源基因二次侵入时,在CRISPR 序列上游前导基因的调控下转录出两种产物反式激活crRNA(Transactivating CRISPR RNA,tracrRNA)和pre-crRNA(pre-CRISPR-derived RNA)。pre-crRNA 和tracrRNA 存在一段互补序列,形成双链区域。随后tracrRNA 指导RNase Ⅲ和Cas9 蛋白对pre-crRNA 剪切,形成成熟的crRNA-tracrRNA-Cas9复合体。该复合体在PAM(Protospacer adjacent motif)位点附近切割crRNA 互补的外源基因序列,从而保护机体免于受到病原的侵入[4-5]。研究者利用这一机制将crRNA 和tracrRNA 的关键基序融合成单一指导RNA(sgRNA),可引导Cas9 蛋白正确识别并切割特定序列,为后续CRISPR-Cas9 基因编辑技术广泛应用奠定基础[6]。
近年来CRISPR-Cas9 基因编辑技术迅速发展,实现了其在多种细胞和动物体内的基因编辑[7-8]。2014 年,首个覆盖人类全基因组的CRISPR-Cas9 敲除文库(GeCKO)构建成功[9]。随后多个研究团队和实验室利用CRISPR-Cas9 敲除文库(GeCKO)进行大规模的遗传筛选,比如利用功能性缺失型策略筛选参与人类免疫缺陷病毒(Human immunodeficiency vi⁃rus,HIV)和甲型流感病毒(Influenza A virus,IAV)等病毒复制阶段的宿主蛋白及介导未整合鼠白血病病毒(Murine leukemia virus,MLV)沉默的宿主蛋白[10-12]。筛选研究的第一步是将sgRNA 慢病毒文库导入到表达Cas9 的细胞中,从而构建全基因组沉默的敲除细胞文库。因此,选择敲除效率高的细胞系是保证获得高覆盖率敲除细胞文库的关键。本研究利用慢病毒感染的方式,将Cas9 基因稳定导入HeLa 细胞中,建立表达Cas9 蛋白的单克隆细胞株。再利用同时表达GFP 和GFP sgRNA 的慢病毒感染HeLa-Cas9 单克隆细胞系,经嘌呤霉素培养液筛选一周后利用流式细胞术检测表达绿色荧光蛋白细胞的百分比以鉴定HeLa-Cas9 单克隆细胞系的敲除效率[13]。最终筛选到一株高敲除活性且细胞活性无显著变化的HeLa-Cas9 单克隆细胞系,为进一步构建基因敲除细胞文库奠定基础。
1 材料与方法
1.1 细胞及质粒 人胚胎肾细胞HEK293T 细胞和宫颈癌细胞系HeLa 细胞株、质粒lentiCRISPR v2-Hyg(潮霉素抗性)、pMD2.G 和pSPAX2 均由本实验室保存;pXPR-011 质粒(Addgene,货号59702,该质粒含GFP 和GFP sgRNA 序列)由中国农业科学院哈尔滨兽医研究所赵东明研究员惠赠。
1.2 主要试剂 质粒提取试剂盒购自Invitrogen 公司;DH5α 感受态细胞购自天根生化科技(北京)有限公司;磷酸钙转染试剂(140 mmol/L NaCl、1.5 mmol/L Na2HPO4·2H2O、50 mmol/L Hepes、pH=7.05)、细胞裂解液(50 mmol/L Hepes、50 mmol/L KCl、0.25% NP-40、1 mmol/L DTT、100 mmol/L Na⁃Cl、pH=7.9)由本实验室配置;胎牛血清购自Wi⁃sent 公司;潮霉素(Hygromycin B Gold)购自InvivoGen公司;兔抗Flag 多克隆抗体和鼠抗β-actin 单克隆抗体(MAb)购自Sigma 公司;红外荧光标记的山羊抗兔IgG(H+L)和红外荧光标记的山羊抗鼠IgG(H+L)IgG 购自KPL 公司;CCK-8 试剂盒购自上海碧云天生物生物技术有限公司。
1.3 稳定表达Cas9 蛋白的细胞系的构建及鉴定
1.3.1 表达Cas9 蛋白慢病毒的制备 将3×106个HEK293T 细胞接种于10 cm 的细胞培养皿中,细胞密度达到30%~40%时,采用磷酸钙转染法将2 μg pMD2.G、9 μg pSPAX2 和9 μg lentiCRISPR v2-Hyg 质粒共转染HEK293T 细胞,8 h 后换液,48 h 后收获上清,用0.45 μm 滤膜过滤细胞碎片,所得上清即为表达Cas9 蛋白的慢病毒原液,置于-80℃备用。
1.3.2 稳定表达Cas9 蛋白的HeLa 细胞系的建立将1.5×105个HeLa 细胞接种于6 孔板中,用1.5 mL 表达Cas9 蛋白的慢病毒原液感染细胞,8 h 后换液,培养至48 h,用潮霉素培养液筛选。为获得最佳潮霉素工作浓度,潮霉素培养液的浓度分别为300 μg/mL、400 μg/mL 和500 μg/mL。每隔2 d 换液。待对照孔的细胞全部死亡后,收集存活细胞。利用western blot 鉴定Cas9 蛋白的表达。将这些细胞用流式分选系统分到96 孔板中,培养7 d 后选单克隆细胞,继续培养2 周后,转移至12 孔板,待细胞密度达到80%~90%,一半细胞用western blot 鉴定Cas9 的表达,另一半细胞继续培养,待鉴定成功表达后,扩大培养并置于液氮保存。将该单克隆细胞稳定传至20 代,western blot 检测Cas9 蛋白表达,这些细胞株即为HeLa-Cas9 单克隆细胞系。
1.3.3 稳定表达Cas9 多克隆细胞系和单克隆细胞系的鉴定 收集稳定表达Cas9 蛋白的细胞,以野生型HeLa 细胞为空白对照,用300 μL 蛋白裂解液裂解细胞,进行SDS-PAGE 凝胶电泳,转印至PVDF膜,5%脱脂乳封闭2 h,以兔抗Flag(1 1 000)多克隆抗体为一抗,羊抗兔IgG(1 10 000)抗体为二抗,检测目的蛋白的表达。
1.4 表达GFP和GFP sgRNA慢病毒的制备及病毒滴度的测定 将HEK293T 细胞按3×106个接种于10 cm细胞培养皿,16 h~18 h 后用磷酸钙转染法将2 μg pMD2.G、9 μg pSPAX2 和9 μg pXPR-011 3 质粒共转染,8 h 换液,48 h 收集上清,用0.45 μm 的滤膜过滤细胞碎片,所得上清即为同时表达GFP 和GFP sgRNA 的 慢 病 毒 原 液, 置 于-80 ℃备 用。 取1 mL HeLa 细胞(1×105个)与1 mL 同时表达GFP 和GFP sgRNA 慢病毒稀释液(2 倍倍比稀释:50 μL~800 μL)混合。慢病毒感染48 h 后,弃去细胞培养液,用PBS 清洗一遍,分别在每孔中加入300 μL 0.25%胰蛋白酶消化5 min,之后加入700 μL 培养液混匀后置于流式管中,利用流式细胞分析仪分析表达绿色荧光蛋白细胞的百分比,根据表达绿色荧光蛋白细胞的百分比为10%~20%的病毒稀释液倍数计算病毒滴度。病毒滴度计算公式:病毒滴度(TU/mL)=(初始感染细胞数×GFP 细胞百分比)/(每孔感染病毒体积)×1 000。
1.5 稳定表达Cas9 蛋白的HeLa 单克隆细胞系中Cas9 敲除活性的检测 将稳定表达Cas9 蛋白的HeLa 细胞系按1×105个/孔接种于6 孔板,感染MOI 0.4 同时表达GFP 和GFP sgRNA 的慢病毒。48 h 后,更换含1 μg/mL 嘌呤霉素的培养液,之后每2 d更换一次含1 μg/mL 嘌呤霉素培养液,同时观察绿色荧光蛋白的表达情况。筛选一周后利用流式分析仪检测表达绿色荧光蛋白细胞的百分比,计算敲除效率%KO=[GFPWT-GFPCas9]/GFPWT(WT:野生型HeLa细胞)。
1.6 稳定表达Cas9 蛋白的HeLa 单克隆细胞系的细胞活性检测 用400 μg/mL 的潮霉素培养液培养HeLa-Cas9 单克隆细胞系7 d 后,以1×104个/孔接种于96 孔板中,每组设置3 个复孔。培养至细胞贴壁,每孔更换预混培养液(其中组成为:10 μL CCK-8 试剂和90 μL 含10% FBS DMEM 培养基),以加了相应量培养液和CCK-8 溶液但没有加入细胞的孔作为空白对照,2 h 后测定OD450nm值。
1.7 二次单克隆化的细胞敲除效率和细胞活性检测经方法1.4 和1.5 鉴定成功的HeLa-Cas9 第8 株单克隆细胞系用流式分选系统再次单克隆化。细胞经过扩大培养后按照方法1.4 和1.5 对其进行Cas9 敲除效率和细胞活性检测。
2 结 果
2.1 稳定表达Cas9 蛋白的细胞系 用lentiCRISPR v2-Hyg 慢病毒感染HeLa 细胞,利用浓度300 μg/mL、400 μg/mL、500 μg/mL 的潮霉素培养液筛选10 d,400 μg/mL 和500 μg/mL 浓度的对照细胞全部死亡。最终选定400 μg/mL 的潮霉素为筛选浓度培养多克隆细胞。Western blot 鉴定Cas9 蛋白表达后,经传至20 代,再次鉴定Cas9 蛋白的表达。结果显示HeLa-Cas9 细胞系在158 ku 出现目的条带,表明Cas9 蛋白在HeLa 细胞中稳定表达(图1A)。上述多克隆细胞经流式分选得到16 株状态良好的单克隆,鉴定结果显示其中有9 株细胞通过western blot 可检测到明显的Cas9 蛋白表达条带,细胞序号分别是2、3、5、7、8、10、11、12 和13,而其余7 株均未检测到Cas9蛋白的表达(图1B),检测正确表达Cas9 的细胞稳定传代20 代,仍能检测到Cas9 蛋白的表达(图略),表明稳定表达Cas9蛋白的HeLa单克隆细胞系构建正确。
2.2 慢病毒滴度的测定结果 利用稀释的慢病毒感染1×105个HeLa 细胞。在病毒感染48 h 时,以未感染病毒的野生型HeLa 为阴性对照,利用流式分析仪分析HeLa 细胞表达绿色荧光蛋白细胞数(图2A),根据表达绿色荧光蛋白细胞百分比和感染病毒剂量绘制曲线,结果显示表达绿色荧光蛋白细胞百分比在40%以下时,感染率与病毒感染剂量呈线性相关(图2B),表明该病毒剂量下(MOI 0.4),一个病毒颗粒只感染一个细胞。本研究选取病毒量为50 μL时,计算可得病毒滴度为:(1×105×18.1%)/50 μL×1 000=3.62×105TU/mL 即每毫升病毒液中含有3.62×105个具有生物活性的病毒粒子。
2.3 建立的HeLa 单克隆细胞系的Cas9 活性 利用MOI 0.4同时表达GFP和GFP sgRNA的慢病毒,感染1×105个HeLa-Cas9 单克隆细胞系,2 d 后再用嘌呤筛选一周,用流式细胞术检测表达绿色荧光蛋白细胞百分比,计算HeLa-Cas9 单克隆细胞系的敲除效率,结果显示除了第2 株细胞以外,其余8 株细胞的敲除效率均在50%以上,其中第8 株和第10 株细胞敲除效率最高,分别为79%和81%(表1),并将这两株细胞记为HeLa-Cas9_8 和HeLa-Cas9_10。倒置荧光显微镜观察HeLa 细胞的绿色荧光蛋白变化,结果显示嘌呤霉素筛选至第6 d时,野生型HeLa 细胞中GFP蛋白表达最多(图3A),此时HeLa-Cas9_8细胞中GFP蛋白表达很少(图3B),表明HeLa-Cas9_8细胞中Cas9蛋白与GFP sgRNA结合靶向GFP基因。
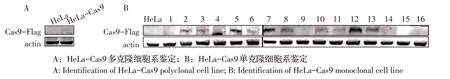
图1 Cas9 蛋白在HeLa 细胞中表达的western blot 鉴定Fig. 1 Identification of Cas9 protein expressed in HeLa cells by western blot
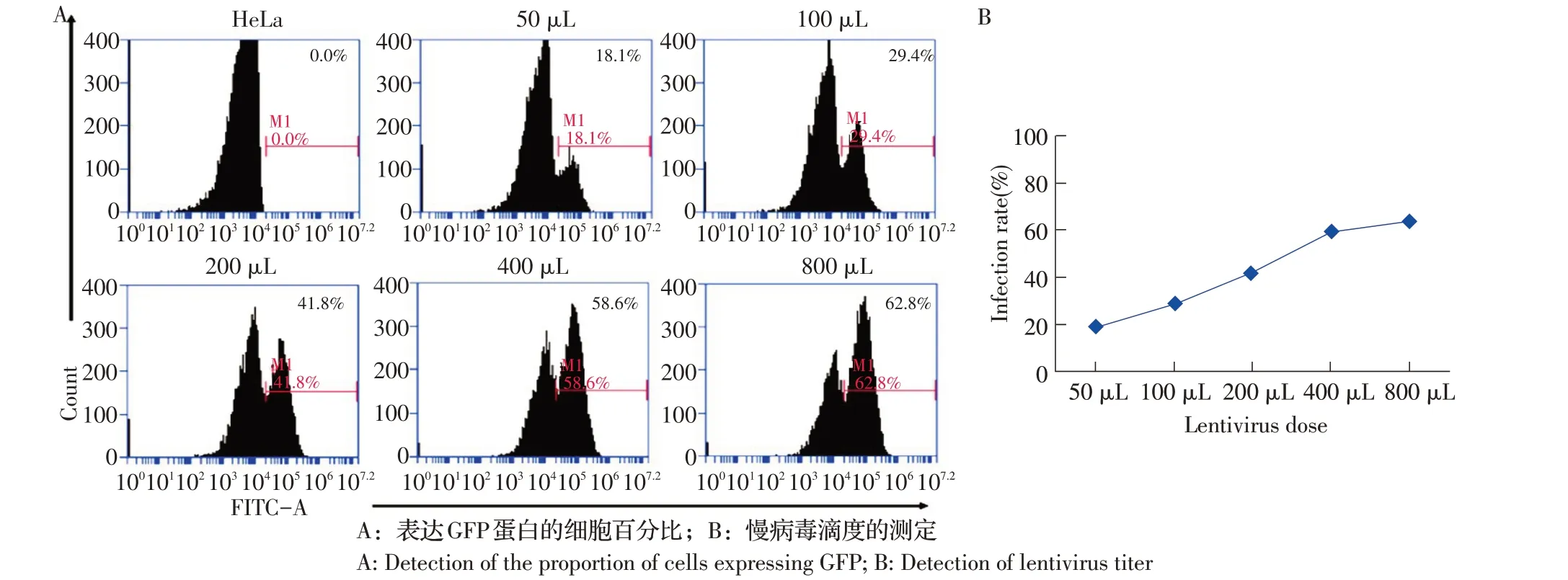
图2 表达GFP 蛋白的细胞百分比的流式细胞术鉴定Fig. 2 Detection of the percentage of HeLa cells expressing green fluorescent protein by flow cytometry

表1 HeLa-Cas9 细胞敲除效率结果Table 1 Knockout efficiency of HeLa-Cas9 polyclonal cells

图3 pXPR-011 慢病毒感染细胞HeLa/HeLa-Cas9_8 后GFP 表达情况Fig. 3 GFP expression in HeLa/HeLa-Cas9_8 cells infected with pXPR-011 lentiviral particles
2.4 稳定表达Cas9 蛋白的HeLa 细胞的细胞活性将野生型HeLa、HeLa-Cas9_8 和HeLa-Cas9_10 细胞系按照1×104个细胞铺于96 孔板,24 h 后每孔加入10 μL CCK-8 试剂,继续培养2 h 后在酶标仪下测OD450nm值,结果显示HeLa-Cas9_8 的细胞活性与野生型HeLa 相比无差异,HeLa-Cas9_10 的细胞活性与野生型HeLa 相比差异显著(图4),表明稳定表达Cas9 蛋白对HeLa 细胞的活性无影响。
2.5 HeLa-Cas9 单克隆细胞系二次单克隆化的细胞敲除效率和细胞活性 将HeLa-Cas9_8 细胞用流式分选系统再次单克隆化,获得9 株单克隆细胞,两周后经过扩大培养后按照方法1.4 和1.5 对其进行Cas9 敲除效率和细胞活性检测。结果显示其中第6株细胞的敲除效率最高为87%(表2),表明二次分选后,表达Cas9 蛋白单克隆细胞之间敲除效率无明显差异。HeLa-Cas9_8-6 细胞活性与野生型HeLa 细胞相比差异不显著(图5),表明稳定表达Cas9 蛋白对HeLa 细胞的活性无影响,HeLa-Cas9_8-6 可作为构建敲除文库的候选细胞系。

图4 表达Cas9 蛋白对细胞活性影响的检测结果Fig. 4 Detection results of the effect with overexpression of Cas9 protein on cell viability western blot

表2 HeLa-Cas9_8 单克隆细胞系敲除效率结果Table 2 Knockout efficiency with HeLa-Cas9_8 monoclonal cells
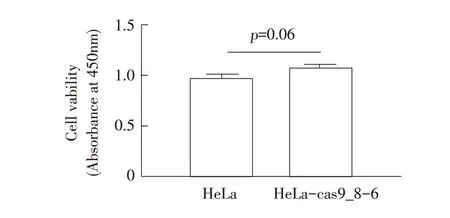
图5 表达Cas9 蛋白对细胞活性影响的检测结果Fig. 5 Detection results of the effect of overexpression of Cas9 protein on cell viability
3 讨 论
21 世纪初人们成功完成人类全基因组测序,为人类疾病相关研究提供了重要信息。为了鉴定某种或多种基因在疾病中发挥的功能,最直观的方法是破坏基因的正常表达并分析其表型[14]。目前,人们可通过RNAi 全基因组筛选,单倍体细胞筛选和CRISPR/Cas9 全基因组筛选这3 种方法实现筛选影响特定表型的未知基因。其中CRISPR-Cas 系统的优势是在基因组中进行编辑,既不会出现RNAi 筛选中脱靶导致的假阳性率和未能完全抑制基因表达而产生的假阴性率,也不受到单倍体细胞筛选技术的细胞严苛限制。CRISPR-Cas9 属于CRISPR-Cas 系统的第Ⅱ类编辑系统,即只需要Cas9 蛋白和sgRNA 便可完成基因靶向编辑,因此得到广泛应用。CRISPR 基因编辑技术由于其设计简单、操作方便和基因编辑效率高等优点已用于包括病毒、肿瘤等多种研究中。研究人员改良并构建了可以产生更高滴度的病毒载体、单载体系统(lentiCRISPR v2、Cas9 和sgRNA 在一个载体中)和双载体系统(Cas9 和sgRNA分别在两个载体中)。单载体系统和双载体系统包装慢病毒的病毒滴度均比lentiCRISPR v1 高,其中双载体系统的病毒产量要高出近100 倍[15]。单载体可能适用于原代细胞筛选或者活体实验,而双载体可以用于各种体外细胞系。本研究利用双载体系统首先构建了表达Cas9 的HeLa 细胞系,通过筛选高敲除效率的细胞系,保证后续导入单条sgRNA 或GeCKO 文库的敲除效果。
本研究意在建立稳定表达Cas9 的HeLa 细胞系,利用GeCKO 双载体文库建立敲除细胞文库。为保证后续构建细胞文库的高覆盖率及良好均一性,筛选遗传背景一致的高切割效率的HeLa-Cas9 单克隆细胞系至关重要。为保证单克隆细胞株严格来源于一个细胞克隆,本研究利用流式分选每个孔一个细胞,培养7 d 后镜下观察确定单克隆株。本实验共分选了16 株单克隆细胞株,但有个别细胞没有检测到Cas9 蛋白的表达[16]。因此,本研究选择了9 株有明显条带的细胞株进行了敲除效率的测定。本研究在测定Cas9 敲除效率时,以MOI 0.4 同时表达GFP 和GFP sgRNA 的慢病毒接种细胞,通过荧光观察和流式检测,结果显示HeLa-Cas9_8 和HeLa-Cas9_10 的敲除效率在抗性筛选第7 d 时达到80%左右。虽然HeLa-Cas9_2 中Cas9 表达很好,但切割效率却未达到50%,推测很有可能与插入的位点有关。本研究通过CCK-8 检测HeLa-Cas9_8 和HeLa-Cas9_10 的细胞活性,结果显示与野生型HeLa 细胞相比,HeLa-Cas9_10 细胞活性显著提高,HeLa-Cas9_8 与野生型HeLa 细胞无显著差别。因此,本研究选择HeLa-Cas9_8 为建立文库的候选细胞株。为严格保证细胞株的遗传背景一致,本研究又将HeLa-Cas9_8 进行了一次单克隆化,获得HeLa-Cas9_8-6细胞株为最终细胞株。进一步通过第二次克隆获得的细胞株的敲除效率与HeLa-Cas9_8 相比并无显著差异,表明经过单轮的流式分选技术便可达到单克隆的目的。本研究获得了一株高敲除效率HeLa-Cas9 单克隆细胞系,该细胞系可用于CRISPR 敲除文库的转导及筛选,为进一步开展HIV-1 等病毒相关宿主基因的筛选及功能鉴定研究奠定基础。
猜你喜欢
湘潮(上半月)(2022年7期)2022-12-06
昆明医科大学学报(2021年8期)2021-08-13
昆明医科大学学报(2021年3期)2021-07-22
猪业科学(2021年3期)2021-05-21
昆明医科大学学报(2021年2期)2021-03-29
中国现代医药杂志(2020年10期)2020-12-14
幽默大师(2020年10期)2020-11-10
中西医结合肝病杂志(2020年2期)2020-10-27
中华诗词(2019年1期)2019-11-14
医药前沿(2019年7期)2019-01-05
