
硅钨基离子液体催化剂的合成及催化氧化脱硫研究
2020-11-08 10:10贺建宏郑长征丁涛郭杨昭
应用化工 2020年10期
贺建宏,郑长征,丁涛,郭杨昭
(西安工程大学 环境与化学工程学院,陕西 西安 710048)
对燃油脱硫的主要措施有加氢脱硫、氧化脱硫、萃取脱硫、吸附脱硫等[1-6],其中氧化脱硫中催化双氧水(H2O2)氧化脱硫为公认的一种极具潜力的脱硫方式[7]。催化剂以有机酸、单体杂多酸、分子筛、真菌生物、离子液体、固体超强酸为主[8-12]。其中离子液体以催化反应温度低、萃取硫容量大、重复性好成为研究热点。硅钨酸催化剂不含磷元素更容易达到环保要求,但催化效率较低,因此开发高效高选择性氧化的硅钨酸类催化剂具有重要意义。
本研究通过硅钨酸与离子液体沉淀反应后生成固体催化剂,使用DMF溶解制备成催化氧化脱硫催化剂溶液。考察处理具有代表性的含硫化合物二苯并噻吩[13]在不同条件下的脱除效果,探讨催化脱硫表观活化能及机理。
1 实验部分
1.1 试剂与仪器
硅钨酸水合物、30% H2O2、N,N-二甲基甲酰胺、四氯化碳、正辛烷均为分析纯;溴化1-苄基-3-甲基咪唑、二苯并噻吩、苯并噻吩均为化学纯。
DF-101S集热式恒温加热磁力搅拌器;DHG-9123A自动程控烘箱;ZDS-2000A型荧光硫测定仪;移液枪(100~1 000 μm);SHB-ⅢA循环水式真空泵;FA1104N电子分析天平;NEXUS 870红外光谱仪。
1.2 催化剂制备
称取1.265 7 g溴化1-苄基-3-甲基咪唑溶于50 mL去离子水中,标记为溶液A;称取3.597 7 g硅钨酸溶解于12.5 mL去离子水,标记为溶液B。将溶液B倒入溶液A中,室温下搅拌1 h。过滤洗涤除去溴化氢,在353 K下烘12 h。将固体研磨得到催化剂。
1.3 模型油的制备
1.3.1 DBT模拟油的制备 称取287.322 6 mg DBT,使用150 mL正辛烷溶解转移至250 mL容量瓶中,室温下用正辛烷定容至刻度线,摇匀制得200 mg/L的含硫模型油Ⅰ。
1.3.2 BT模拟油的制备 称取261.578 0 mg BT,使用150 mL正辛烷溶解转移至250 mL容量瓶中,室温下用正辛烷定容至刻度线,摇匀制得200 mg/L的含硫模型油Ⅱ。
1.4 脱硫实验
以处理DBT模拟油Ⅰ为例。在25 mL单口烧瓶中加入一定量催化剂用DMF将催化剂充分溶解,再加10 mL DBT模拟油升温至目标温度并磁力搅拌回流冷凝,加入30% H2O2反应2 h,期间每隔15 min取上层清油过滤,检测油相含硫量并记录实验结果。
1.5 硫含量检测
依据中华人民共和国石油化工行业标准SH/T 0689—2000轻质烃及发动机燃料和其他油品的总硫含量测定法(紫外荧光法)所提供的检测方法使用荧光硫测定仪检测。检测原理:1 050 ℃富氧条件中,硫被氧化成二氧化硫,二氧化硫吸收紫外光的能量转变为激发态的二氧化硫,当激发态的二氧化硫返回稳态的二氧化硫时发射荧光,由检测器所得信号值计算出试样的硫含量。
设备检测条件:室温下开机检验,液态进样,进样速度30 μL/min,高压460 Pa,燃烧室1 050 ℃,氩气120 mL/min,裂解氧480 mL/min,进口氧100 mL/min,进样量10 μL,检测完成后间隔时间3 min再次进样。自动显示硫含量,每个样检测3次取平均值。
2 结果与讨论
2.1 催化剂红外光谱(FTIR)分析
图1a为溴化1-苄基-3-甲基咪唑的红外吸收谱图,图1b、1c分别为催化剂、使用后催化剂的红外谱图。
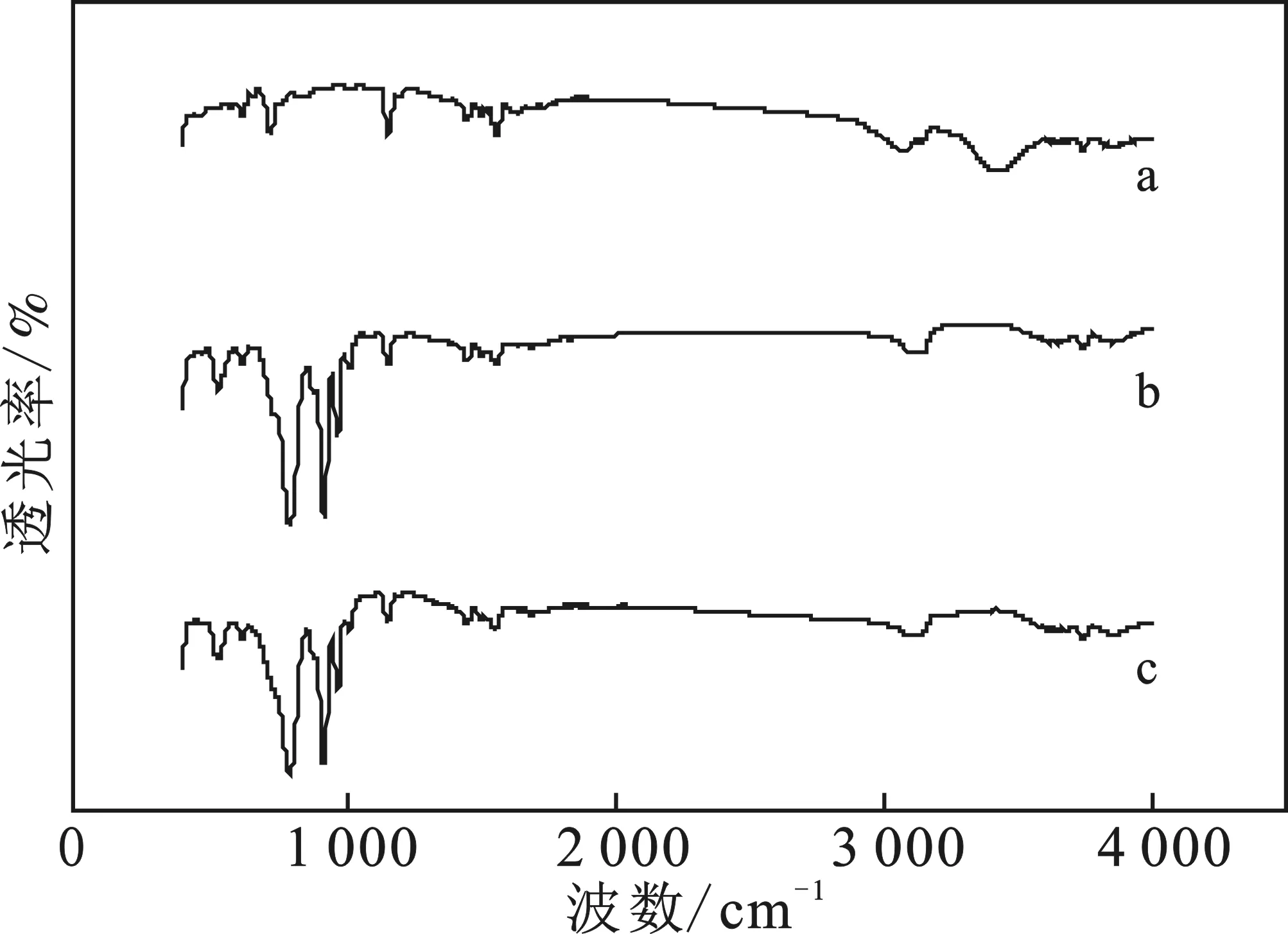
图1 催化剂的红外光谱图Fig.1 FTIR spectra of the catalysis
由图1a可知,图中3 076 cm-1为苯环碳氢伸缩振动吸收峰,1 567 cm-1为苯环骨架振动吸收峰,727 cm-1为苯环上取代基吸收峰,3 429 cm-1为分子内N—H形成的氢键吸收峰,1 158 cm-1为咪唑环伸缩振动吸收峰。由图1b、1c可知,谱图中921 cm-1吸收峰增强为苄基亚甲基C—H吸收峰[14],792,921 cm-1为硅钨杂多酸特征吸收峰。催化剂制备过程中离子液体的特征峰均能检测到,表明制得含离子液体的固体杂多酸催化剂。
2.2 催化剂XRD分析
图2是硅钨酸水合物(a)制备所得催化剂(b)的XRD谱图。
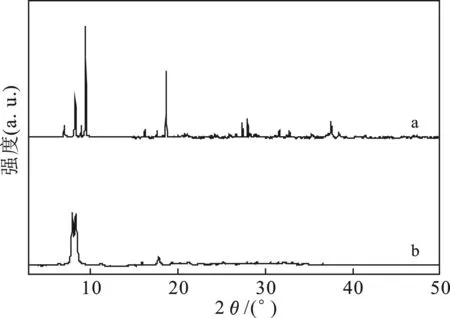
图2 催化剂的X射线衍射谱图Fig.2 XRD patterns of the catalysis
由图2a可知,2θ=7.00,8.29,17.78°处的衍射峰是硅钨阴离子产生的。由图2b可知,2θ= 8.23,17.59°处为硅钨阴离子衍射峰,且两处衍射峰均变宽,其他位置特征衍射峰消失,表明形成较小其他类型晶体,其他衍射峰缺失可能为离子液体的影响。
2.3 催化剂TG-DTG分析
为验证硅钨基杂多酸在制备过程是否被破坏,对催化剂做TG-DTG分析,结果见图3。
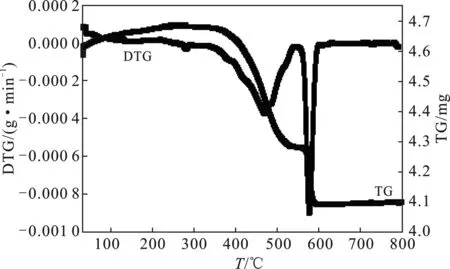
图3 催化剂的热重图Fig.3 TG-DTG characterization of the catalysis
由图3可知,在343 ℃之前催化剂有少许损失,是由于催化剂中水分蒸发所致。343 ℃之后催化剂开始分解,硅钨阴离子和咪唑阳离子上乙烯链共同分解导致失重,594 ℃之后主要由于咪唑环的分解,导致失重。结合以上催化剂随着温度的分解过程可知,硅钨阴离子和咪唑阳离子在制备过程中没有被破坏。
2.4 不同体系对DBT的脱除效果
反应条件:353 K,50 mg催化剂,V(DMF)=2 mL,V(H2O2)=2 mL,V(模拟油Ⅰ)=10 mL,t=8 h 时,不同反应体系下的脱除效果见表1。
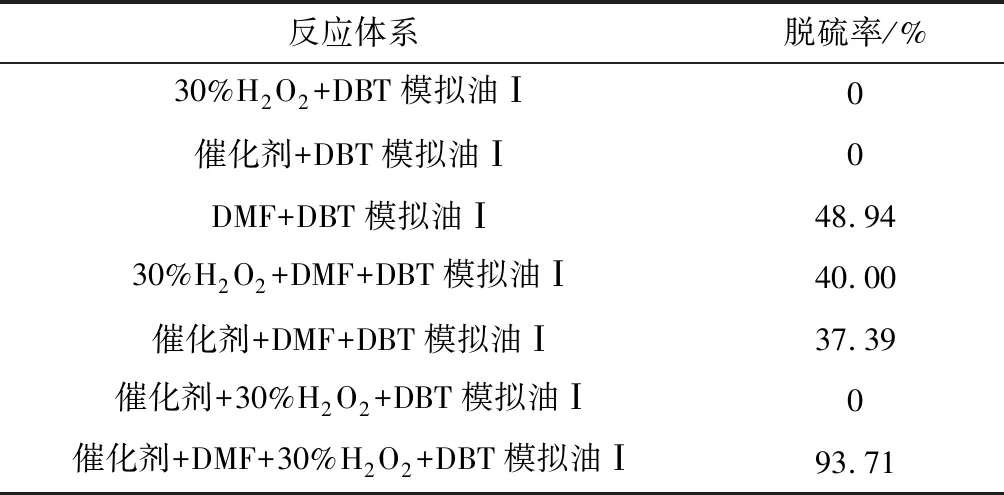
表1 不同反应体系下的脱除率Table 1 Sulfur removal rate in different reaction systems
由表1可知,无论单独使用或混合使用催化剂、H2O2时均无催化效果,加入DMF后催化脱硫效果提升至93.71%比DMF单独萃取效果48.94%有很大增加,但是当催化剂与DMF连用时会降低DMF的萃取能力。DMF在反应中充当催化剂溶剂及萃取作用。同时使用催化剂、H2O2、DMF形成了萃取耦合催化氧化体系,达到了很好的催化氧化脱硫效果。
2.5 不同反应条件对脱硫效果的影响
图4a为反应温度对DBT脱除反应的影响,反应条件:m(催化剂)=40 mg,V(DMF)=2 mL,V(H2O2)=2 mL,V(模型油Ⅰ)=10 mL。
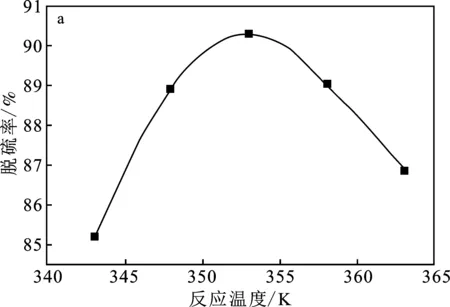

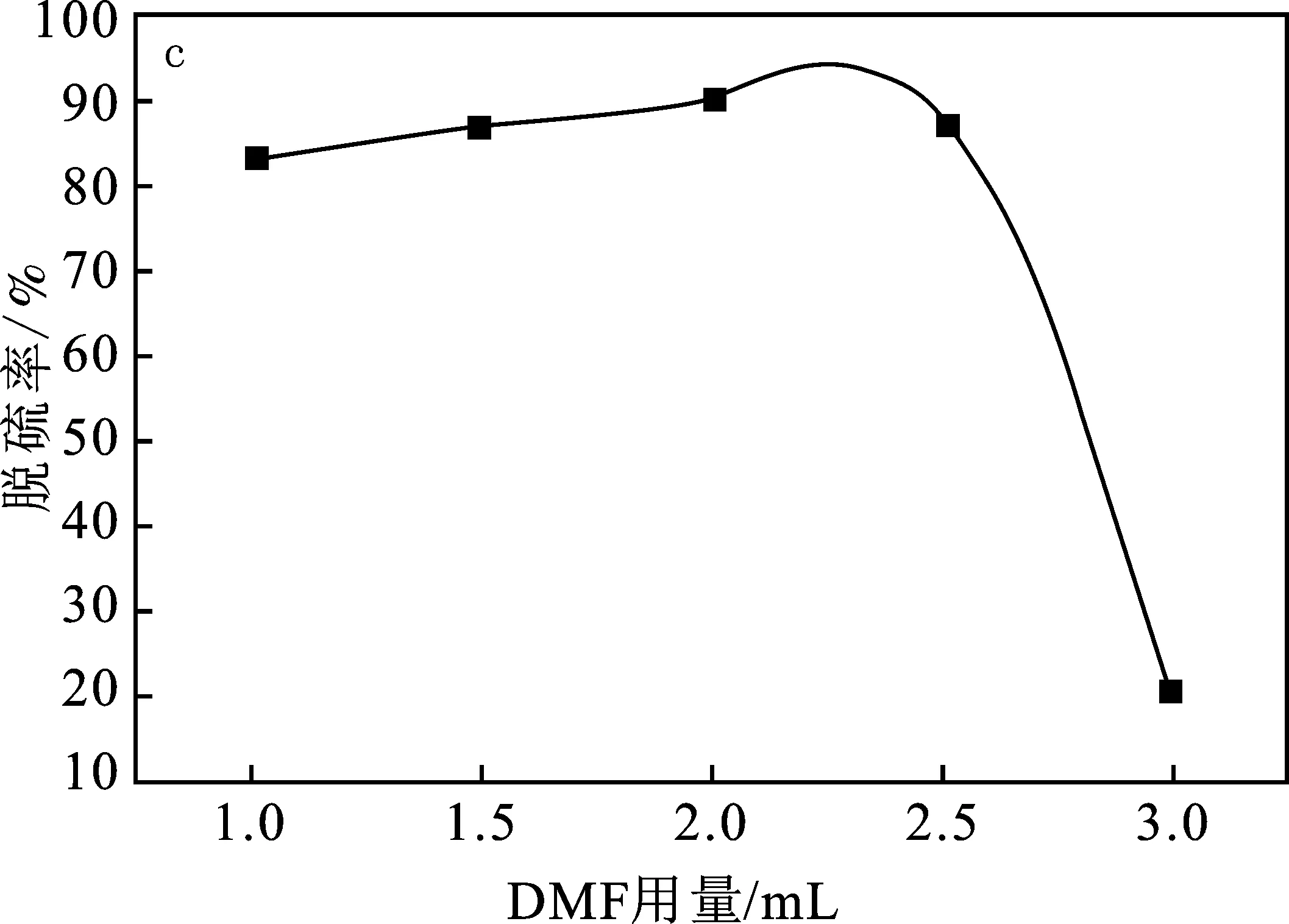

图4 不同反应条件下的脱除率Fig.4 Sulfur removal rate in different reaction conditions
由图4a可知,DBT模拟油的去除效果随着反应温度的升高呈现先升高后降低的趋势。这是因为温度在353 K后H2O2分解剧烈,产生大量水使催化剂部分析出,导致脱硫效果降低,而活性中心随着反应温度升高变好,因此该体系在353 K时效果达到最佳。故后续实验均在353 K反应。
图4b为催化剂用量对反应的影响,反应条件:T=353 K,V(DMF)=2 mL,V(H2O2)=2 mL,V(模型油Ⅰ)=10 mL。
由图4b可知,脱硫率随着催化剂用量增加,催化效果呈现先增大后降低的趋势。这是因为催化剂溶解于DMF量>20 mg/mL后会降低DBT的溶解,导致传质作用进入DMF中的DBT减少,降低催化效果。在20 mg/mL浓度之前随着催化剂用量增加催化效果增加,原因在于DMF中DBT溶解较多,催化剂浓度的增加使有效碰撞增多。在40 mg时表现出最佳的催化效果,故后续实验均使用40 mg催化剂。
图4c为DMF用量对脱硫率的影响反应条件:T=353 K,m(催化剂)=40 mg,V(H2O2)=2 mL,V(模型油Ⅰ)=10 mL。
由图4c可知,随着DMF用量增加,催化剂浓度降低,浓度达到20 mg/mL时达到最佳催化效果,并且催化效果出现拐点,稀释至20 mg/mL以后呈断崖式下降,得出20 mg/mL的催化剂催化效果最佳。原因在于催化剂被稀释后单位时间内形成氧化催化剂的过渡形态分子数减少,导致催化效果降低。后续实验使用2 mL浓度20 mg/mL的催化体系。
图4d为氧化剂用量对脱硫率的影响,反应条件:T=353 K,m(催化剂)=40 mg,V(DMF)=2 mL,V(模型油Ⅰ)=10 mL。
由图4d可知,随着H2O2用量增大催化脱硫效果呈递增趋势,在3 mL时脱硫效果达到最大94.01%,随着H2O2用量继续增加,H2O2分解产生的水萃取DMF导致催化剂析出,降低脱硫率。但3 mL使用量时O/S为386,总体O/S的值较大原因在于催化剂与H2O2的接触性不好。
在最佳反应条件T=353 K,m(催化剂)=40 mg,V(DMF)=2 mL,V(H2O2)=3 mL,V(模型油Ⅰ)=10 mL重复脱硫实验得出该体系脱硫率为90.28%。
2.6 催化剂对不同含硫物的催化效果
反应条件:40 mg催化剂,V(DMF)=2 mL,V(H2O2)=3 mL,V(模拟油Ⅰ)=10 mL时,不同温度下DBT、BT的脱除效果见图5。

图5 不同温度下DBT、BT的脱除效果Fig.5 DBT,BT removal effect at different temperatures
由图5可知,催化剂对DBT的催化氧化效果大于对BT的催化氧化效果,但对BT的催化氧化随着反应温度的升高呈上升的趋势与催化氧化DBT趋势不同,即对含硫物的催化氧化有一定的选择性。引起这种差异的主要原因在于,DBT的硫原子电子云密度5.578大于BT的硫原子电子云密度5.568,使得DBT的氧化较BT容易,因此该体系下脱硫效果DBT>BT。与侯良培等[15]催化氧化脱硫的研究结果一致。
2.7 催化剂的回收
为确定催化剂反应前后是否发生结构变化,反应完成后分离下层液体,加入等体积水混合摇匀后有白色固体析出,过滤所得即为催化剂,用红外光谱分析其中官能团是否发生变化。由图1可知,催化剂反应前后官能团未发生变化。
2.8 催化反应的动力学分析
2.8.1 反应动力学常数 不同温度下DBT含量随时间的变化见图6。反应条件:40 mg催化剂,V(DMF)=2 mL,V(H2O2)=3 mL,V(模拟油Ⅰ)=10 mL。

图6 不同温度下DBT含量随时间的变化Fig.6 Changes of DBT content with time at different temperatures
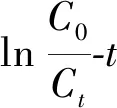

图7 不同温度下催化氧化DBT的反应速率Fig.7 Reaction rate of catalytic oxidation of DBT at different temperatures
C0为初始含硫量,Ct为tmin时含硫量。
(1)
(2)
做定积分得:
(3)
做不定积分得:
lnCt=-kt+常数
(4)
通过对lnCt-t做线性拟合,得出直线的斜率即为反应速率常数k。由图7可知,随着温度的升高,反应速率常数增大,反应速率由343 K的0.017 09 min-1增至363 K的0.026 56 min-1,反应速率加快与监测的反应曲线一致。
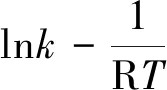
(5)
对其取对数得:
(6)


图8 343~363 K温度下的线性拟合 linear fitting at 343~363 K
2.9 反应历程
由图9可知,该反应体系中随着温度的升高,反应速率增大,但348 K后增速减缓是由于氧化反应过程中水的产生导致催化剂部分析出,同时高浓度催化剂影响DBT在油与DMF水混合相间的转移。
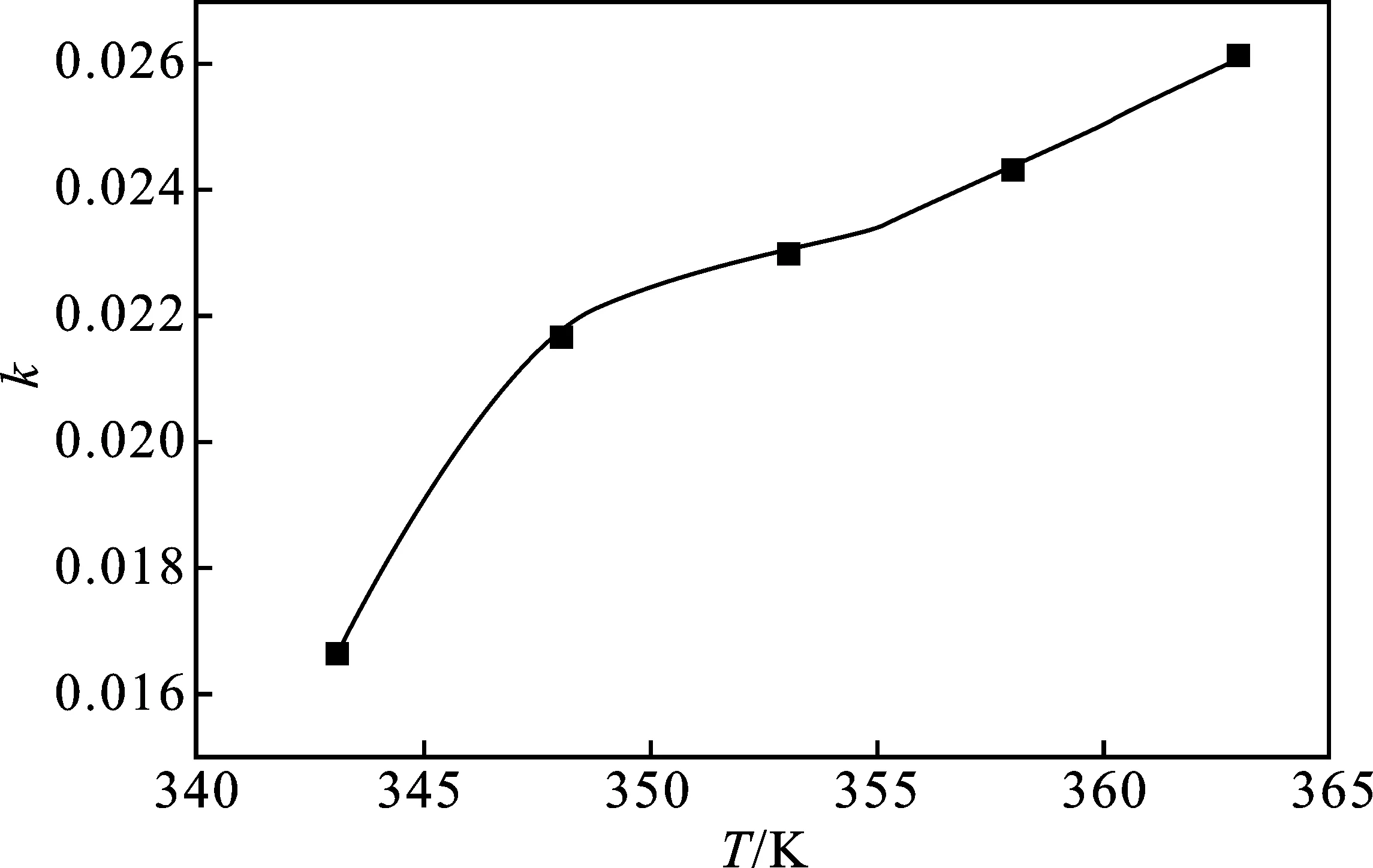
图9 最佳反应条件下k-T关系Fig.9 k-T relationship diagram under optimal reaction conditions
由于催化剂大部分溶解于DMF可以排除DBT在DMF水混合相中扩散对反应的影响,反应过程推断为DBT萃取至DMF水混合相、DBT被氧化两个过程,主要影响集中于萃取过程,结合之前分析工作随着温度升高氧化剂分解加快,反应过程产生的水萃取DMF导致催化剂部分被析出,降低了催化效果与DMF的萃取效果。
2.10 氧化产物的红外表征
反应完成后分离油相,使用四氯化碳体积比为1∶1萃取下层混合相,除去四氯化碳,析出淡黄色的晶体,使用红外光谱检测晶体及DBT,结果见图10。

图10 DBT和氧化物红外光谱Fig.10 DBT and oxide infrared spectrum
由图10可知,红外谱图中产物出现硫氧双键吸收峰。图中1 163,1 288 cm-1为二苯并噻砜的硫氧双键吸收峰,1 046 cm-1处有少量的二苯并亚砜的硫氧双键吸收砜。可以判断DBT经催化剂催化氧化生成物硫原子上携带1~2个不等的氧原子。
2.11 氧化脱硫过程
综上所述,反应过程为DBT从模拟油萃取至DMF和H2O2混合相中,混合相中过氧化氢将催化剂氧化成过氧化形式,由于催化剂使用前已成均一态,所以,DBT在DMF中的传质阻力为零,反应阻力主要为DBT在油、DMF两相间的传质,催化剂被氧化。传质作用到达DMF相的DBT与过氧化的催化剂反应生成砜或亚砜,由于产物的极性较大会保留在水相中,达到降硫的目的。
3 结论
实验成功制得不溶于油与水的固体催化剂,催化剂自身只溶解于DMF,反应完成后只需添加与DMF等体积的水即可析出催化剂,提高催化剂的回收性能,简化回收手段。反应中使用催化剂溶液进行催化反应,使反应由原有的液固液三相变为液液两相接触,反应过程变为单一的液相反应。反应过程中产生的水与DMF混溶导致催化剂析出,因此需要掌控催化剂浓度的同时控制H2O2与DMF的比例。
综上所述,在353 K反应体系中氧硫比为386时,使用2 mL浓度为20 mg/mL催化剂DMF溶液催化氧化DBT,2 h可以达到最佳脱硫效果,脱硫率为90.28%,该体系下氧化二苯并噻吩的表观活化能为21.262 kJ/mol。
猜你喜欢
环球时报(2022-05-23)2022-05-23
金桥(2021年4期)2021-05-21
电子制作(2019年7期)2019-04-25
电子制作(2019年7期)2019-04-25
传感器世界(2019年11期)2019-02-17
西安工程大学学报(2016年3期)2016-06-05
中国卫生标准管理(2015年2期)2016-01-14
陶瓷学报(2015年4期)2015-12-17
西南军医(2015年2期)2015-01-22
天津师范大学学报(自然科学版)(2014年2期)2014-08-06
