
伴有肾损害的十例Fabry病临床及病理特征
2020-11-03 02:03何娟王頔田秀娟于艳许国双
临床肾脏病杂志 2020年10期
何娟 王頔 田秀娟 于艳 许国双
710000 西安,空军军医大学第一附属医院肾脏内科
法布里病(Fabry病)是一种罕见的X染色体隐性遗传的先天性α-半乳糖苷酶A(α-galactosidase A,α-Gal A)缺乏疾病[1]。GLA基因发生突变导致α-Gal A酶的活性降低,引起其作用底物神经鞘脂类化合物(绝大部分为三聚己糖神经酰胺,又称Gb3)在溶酶体中不断蓄积,临床多表现为皮肤血管角质瘤、少汗、角膜病变、听力下降、外周及中枢神经病变、心脏、肾功能受损等多系统受累的经典型;少数有残余酶活性的患者则表现为以心脏和(或)肾脏受累为主、成年发病的迟发型[2]。1898年皮肤科医生Anderson和Johann Fabry各首先报告1例,故名Anderson-Fabry病,简称Fabry病[3]。由于肾脏是Gb3的主要沉积部位和主要靶器官之一,且病情进展至终末期肾病(end stage renal disease,ESRD)是Fabry病患者死亡的主要原因,因此Fabry病肾损害作为Fabry病的重要组成部分备受关注[3]。由于该病较为少见,且缺乏特异性临床表现,因此易被误诊。包括了Fabry病结局调查的366例Fabry病欧洲患者中,从出现症状到明确诊断的平均延迟时间为男性13.7年,女性16.3年[1]。
目前有越来越多的蛋白尿患者和(或)慢性肾脏病(chronic kidney disease,CKD)以及透析患者被确诊为Fabry病的报道[4],提示在蛋白尿和(或)CKD患者中筛查Fabry病的重要性。空军军医大学第一附属医院肾脏内科在1997年首次报告l例Fabry病后[5],至今共收治10例Fabry病。本研究总结了上述患者的临床表现及病理特点,以期提高对此病的认识。
资料与方法
一、研究对象
收集空军军医大学第一附属医院肾脏内科自1997年9月至2020年1月确诊为Fabry病的患者共10例。Fabry病诊断依据:(1)病理超微结构发现细胞内髓样小体,包括肾组织、皮肤组织或神经组织病理检测;(2)α-Gal A酶活性部分或完全丧失;(3)基因检测发现突变;对于男性患者具备上述3条中1条即可诊断,而女性患者具备(1)和(或)(3)可诊断。
二、方法
1.患者资料收集 收集所有患者的临床、实验室及各项特殊检查结果,包括常规体格检查、血常规、尿常规、大便常规、24 h尿蛋白定量、估算肾小球滤过率(estimated glomerular filtration rate,eGFR)及肝功能检查、肾功能检查、心电图、心脏超声,部分患者进行了听力、头颅等检查。对于有肾脏穿刺指征的9例患者进行了超声引导下肾活检术,肾组织病理检查包括光镜、免疫荧光和透射电镜。
2.肾活检病理检查方法 (1)光镜:标本石蜡包埋,切片厚2 μm,分别进行HE、PAS、PASM-Masson、Masson染色。(2)免疫荧光:采用4 μm厚冰冻切片,以异硫氰酸荧光标记的鼠抗人荧光抗体行肾组织冰冻切片直接法染色,检测组织中免疫球蛋白(immunoglobulin,Ig)G、IgA、IgM、补体C3、补体C4、补体C1q。(3)电镜:组织经戊二醛及锇酸双重固定,Epon812环氧树脂包埋,行1 μm半薄切片,经0.5%甲苯胺蓝水溶液染色,光镜下观察;行50~70 nm超薄切片,经醋酸铀、柠檬酸铅双重染色,置JEM-1200Ex透射电镜下观察。
三、统计学方法
结 果
一、患者的一般情况
10例患者的一般特征见表1,患者全部为男性,确诊时年龄范围13~66岁,平均年龄28.6岁。30岁之前发病的患者共7例。患者自述家族史均不详。
二、患者的临床表现
1.肾脏表现 9例患者表现为不同程度的蛋白尿,平均尿蛋白水平1 582 mg/24 h,其中24 h蛋白尿>1 g者共6例,1例患者表现为肾病综合征,24 h尿蛋白定量<1 g共3例。7例患者出现血尿,均为镜下血尿。肾功能受损(血肌酐>133 μmol/L)共5例,7例合并eGPR下降[(76.4±53.0)mL·min-1·(1.73 m2)-1](表1)。1例肾病综合征起病的男性患者,就诊时表现为重度双下肢水肿,且已进展至ESRD。其余患者无明显水肿或高血压。B型超声查双肾体积基本正常。2例患者分别在确诊Fabry病后2年6个月、1年7个月余进展至ESRD。另有2例患者死亡,死亡原因不详。其余5例患者均行CKD二级预防治疗中。
2.肾外表现 发病率最高的并发症为心脏受累(5例),心脏超声造影检查结果提示心脏损害以室间隔增厚为主(3例),1例合并少量二尖瓣反流,1例合并病态窦房结综合征。我中心患者尚无房室传导阻滞、PR间期缩短、ST段和T波的异常等心脏传导功能异常的表现。此外,3例患者合并皮疹,1例合并皮肤血管角质瘤,2例肢端感觉异常,3例听力下降,1例角膜病变,1例反复上腹部不规则疼痛。我中心患者尚无脑血管损害表现的患者。
3.患者的皮肤特征 其中1例患者表现为皮肤血管角化瘤,行皮肤活检(图1)可见浅表肿瘤,位于真皮乳头层,瘤体内可见多数扩张的毛细血管,外衬一层内皮细胞,腔内充满着红细胞,瘤体两侧的表皮突向下延伸,包绕大部分瘤体。

注:红色箭头代表扩张的毛细血管外衬内皮细胞腔内充满的红细胞。图中比例尺为150 μm。图1 患者的皮肤活检病理图 A.HE(40×);B.HE(100×)
4.患者的肾脏病理特征 (1)光镜:易见肾小球节段性硬化(图2A)。部分患者可见肾小球球性硬化,肾小管局灶状萎缩,间质局灶状淋巴和单核细胞浸润伴纤维化(图2B)。所有患者肾小球足细胞呈弥漫性肿胀、空泡变性和泡沫样改变(图2C、图2D)。
(2)免疫荧光:IgM为阴性或弱(+),沿毛细血管袢、系膜区或节段硬化部位沉积。其余免疫球蛋白和补体检测结果均为阴性(图2E)。
(3)电镜:所有患者足细胞内均可见大量特征性的髓样小体,直径0.3~10.0 μm,多数3.0 μm,呈圆形或致密的螺纹状结构,类似“葱皮”或“斑马”样外观,具有明暗相间的条纹(图2F),部分患者系膜细胞和肾小管上皮细胞胞浆内也可见不同数量的髓样小体。
讨 论
Fabry病又称弥漫性躯体血管角化瘤,是一种罕见的X连锁伴性遗传的溶酶体贮积症,其致病基因位于X染色体长臂Xq21.33-Xq22上。其发病机制为编码α-Gal A的基因发生突变,目前已经发现了超过500种突变[4]。通过点突变,其代谢底物Gb3和相关鞘磷脂不能被裂解而在体内血管内皮细胞和其他细胞蓄积,进而在皮肤、神经、心脏、肾脏、肺、胃肠道、眼、耳、骨骼等部位沉积,造成多器官多系统损伤,患者多在40~50岁死于尿毒症或心血管并发症[6]。
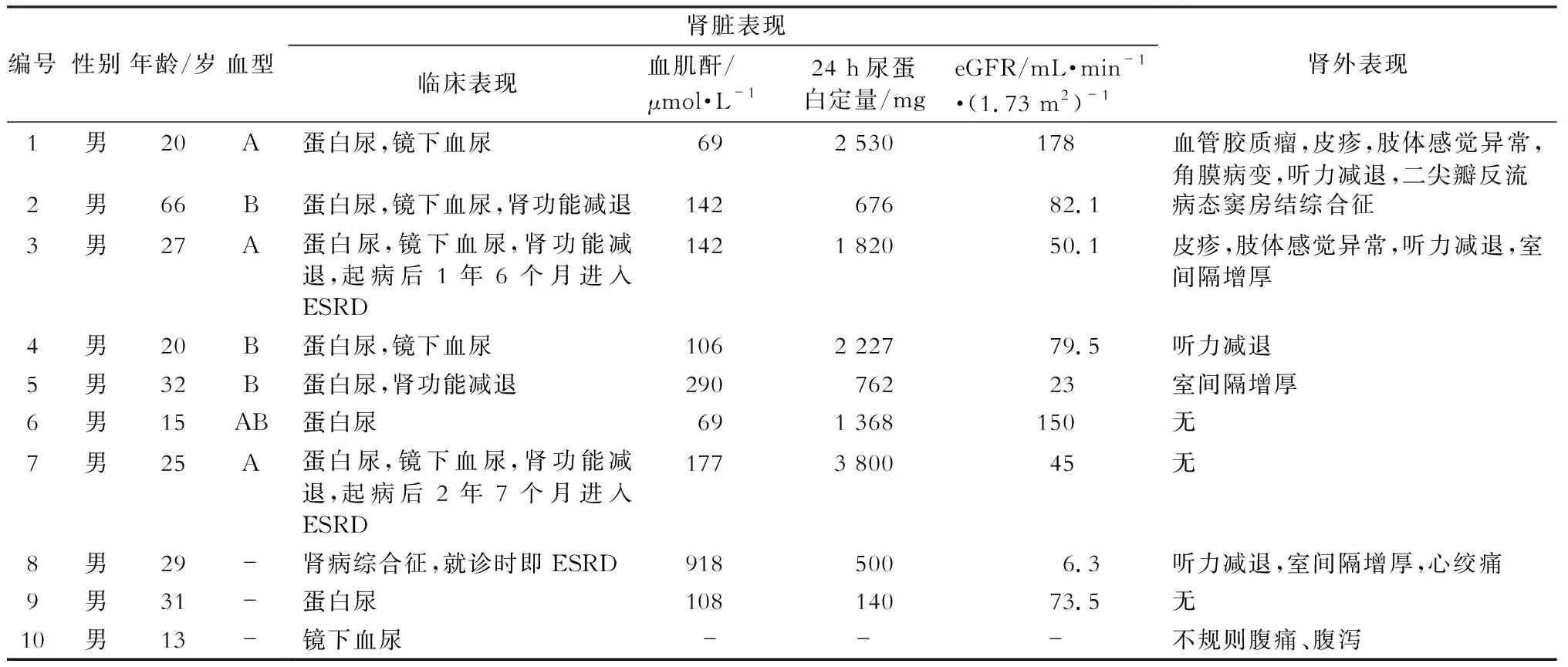
表1 10例Fabry患者的临床特点

注:图A中红色箭头表示肾小球节段性硬化;图B中红色箭头表示肾小球球性硬化,黑色箭头表示肾小管局灶状萎缩;图C黄色框线为扩大后的图D;图D中红色箭头表示肾小球足细胞肿胀,空泡样变;图F中红色箭头表示足细胞内的大量髓样小体。图中比例尺为150 μm。图2 患者的肾活检病理图 A.PAS染色(光镜,400×);B.PASM+Masson染色(光镜,200×);C.PAS染色(光镜,600×);D.PAS染色(光镜,1 000×);E.IgM免疫荧光(400×);F.电镜(4 000×)
既往估计的Fabry病的发病率波动于1∶117 000~1∶40 000[7]。最近的新生儿筛查研究显示,Fabry的患病率明显高于预期,在男性儿童中高达1∶3 100[8],成年男性的发病率1/60 000~1/4 000[9]。本研究报道的我科目前诊断的10例患者均为男性。有研究报道,在行肾脏替代治疗的患者中,0.33%的男性患者、0.10%女性患者患有Fabry病[10]。对于女性的Fabry病目前研究较少,其白细胞α-Gal A水平可从仅低于正常至几乎为零不等。多数杂合子女性患者较少或仅在生命晚期出现轻中度的肾或心脏问题,也可表现为典型的Fabry病。因中国尚缺乏流行病学数据,目前仅有部分病例报道,确诊患者仍以经典型为主,且规范的基因、酶活性检测结果及家系调查报道较少。
该病临床表现多种多样,极易漏诊、误诊。结合患者体内α-Gal A活性水平、受累器官等,可分为两型[11]:(1)经典型表现为α-Gal A酶活性明显下降甚至完全缺失,脑、肾脏、心脏、周围神经等多系统受累;(2)迟发型表现为α-Gal A酶活性部分下降,往往限于心脏或肾脏受累,常无血管瘤、听力损伤、肢端感觉异常、角膜混浊和少汗等早期表现。绝大部分男性患者和极少部分女性患者为经典型,大部分女性患者为迟发型。
文献报道的Fabry病心脏表现有左心室肥厚(52.7%)、存在心脏瓣膜病变(35.4%)、冠状动脉病变、传导异常,最终导致心力衰竭、心律失常与心肌梗死[12]。青年脑卒中患者中,Fabry病占0.5%,高血压合并左心室肥厚占0.9%,特发性肥厚型心肌病中占0.5%~1.0%。40岁以上表现为肥厚型心肌病的男性患者中,约有6.3%最终确诊为Fabry病[13]。本中心患者肾外表现以心脏受累最为突出,但患者均无明显胸闷、气短等临床表现。Sachdev等[14]对153例不明原因、晚期发病的肥厚型心肌病男性患者进行分析,结果显示年龄在40岁以上的患者中,有6.3%为Fabry病所致。因此,对该年龄段的男性患者,当出现难以解释的左心室肥厚时,除外系统性淀粉样变性病、心肌损伤等相关因素,还应考虑患有Fabry病的可能。
Fabry病常见的眼部病变包括角膜营养不良、晶状体混浊和缝隙样改变,眼底检查可发现视网膜血管迂曲。在白种人群中,几乎所有男性患者和70%~90%杂合子女性患者存在眼部病变[14]。然而,日本学者在对230例男性Fabry病患者研究后发现,日本患者中较少出现Fabry病典型的眼部病变[15]。相似的是,本组10例患者中仅1例男性患者存在角膜病变。因此,有待收集更多的病例进一步验证其眼部病变是否与人种相关。
Fabry病患者临床症状与其血型亦有一定联系[16]。AB型和B型血细胞抗原中有需要α-GalA水解的糖鞘脂,其体内糖鞘脂比其他血型者储积要多,因此推测男性尤其是AB和B型血型者患病率较高,且临床症状及病情发展均相对较重。本研究10例患者中共7例行血型鉴定,其中B型患者3人,AB型患者1人。这对判断患者临床症状及病情进展具有一定意义。
肾脏为Fabry病的重要累及器官之一,在Fabry病合并CKD患者中,男性占59.3%,平均年龄为54.81岁,<44岁者占25.7%,45~66岁者占49.8%,>66岁者占24.5%[4]。本研究中患者平均年龄为28.6岁,均为男性。Fabry病肾损伤大致可以分为四期:(1)Gb3的沉积;(2)细胞损伤;(3)器官损伤;(4)肾功能进行性损伤[17]。有报道指出,妊娠17周即可在许多肾细胞类型[18]及胎盘组织中[19]检测到Gb3沉积。在最近的一项儿童随机对照试验中,在所有eGFR正常及尿白蛋白/肌酐<30 mg/g的Fabry病患者,通过肾活检,电镜下均可见Gb3在许多肾细胞中(包括肾小球内皮细胞、系膜细胞、管周上皮细胞、足细胞等)沉积,尤以足细胞受累明显,并可观察到Fabry病特有的动脉病变[20]。随着年龄增长,Gb3在足细胞中沉积逐渐增多,并与肾损伤(白蛋白尿、eGFR下降)呈正相关[21]。
当Fabry病患者出现肾脏表现时,主要表现为蛋白尿和进行性肾功能不全。蛋白尿可能源自肾小管或肾小球,可能始于青少年早期,但更常见于成年早期[22]。蛋白尿水平高[尿蛋白/肌酐比>1500 mg/g]的Fabry病成年男性的肾功能下降也最快;该研究也同时发现,在Fabry病成年女性患者中,蛋白尿与肾功能下降的关系远没有那么密切[23]。因此,蛋白尿本身是Fabry病进展的直接促进因素这一观念可能并不正确。本组患者临床尿液检查多表现为轻、中度的非肾病综合征性蛋白尿,无肉眼血尿,无明显水肿、低蛋白血症和高脂血症,仅1例男性患者以肾病综合征、重度水肿、ESRD就诊。与一般慢性肾小球肾炎相比无明显特异性,很难根据临床表现确定诊断。事实上,早期肾脏损害表现容易被忽视,可表现尿液浓缩障碍,即多尿、夜尿增多、尿比重减低等。在临床出现明显蛋白尿前,尿中足细胞丢失已经开始增加。随病程进展逐渐出现蛋白尿,伴随镜下血尿、肾功能损害、eGFR下降,患者多在30岁左右进入ESRD。本中心进入ESRD的3例男性患者年龄分别为25岁、27岁、29岁。在美国,有12%的Fabry病患者正在接受透析治疗,而有0.25%~1%的慢性维持性血液透析的男性患者在最初漏诊Fabry病[10],但透析和肾移植均不能提高其生存率,患者肾脏损害程度与Fabry病特征性肾外临床表现之间无直接联系,多数患者于40~50岁时因心、脑血管并发症或尿毒症死亡[24]。
肾组织形态学改变有其突出特点,主要表现为光镜下肾小球不同程度局灶节段性硬化,肾小球足细胞和肾小管上皮细胞可观察到数量不等的空泡变性。电镜改变显示细胞溶酶体内可见特征性嗜锇性、同心圆样包涵体,小体高电子密度,圆形或卵圆形,多为1~3 μm,小体内部呈层状,层间距较一致,形似“斑马皮”或“洋葱皮”,故称“斑马小体”、“洋葱皮小体”或“髓鞘小体”。肾活检有助于确定细胞损伤的严重程度,是临床确诊该病和评估肾脏受累程度的重要依据。
除了特征性的临床表现、病理特点,还可对Fabry病患者进行α-Gal A酶活性检测和基因检测[25]。外周血白细胞外周白细胞α-Gal A活性低于25%~30%可确诊,需要注意的是,部分杂合子女性患者或者延迟起病患者的酶可能会是低活性(≤10%)、活性中等降低(20%~40%)或者活性正常,故该酶活性正常者不能除外该病。血和尿中Gb3水平升高,可协助临床诊断,尤其是女性患者,确诊需要进行α-Gal A基因检测。α-Gal A基因突变类型至今已报道了400余种(人类基因突变数据库http://www.hgmd.cf.ac.uk)。近年来,中国学者研究发现在中国人群中已有20余种突变类型被报道[26],相较于组织活检,α-Gal A基因检测作为Fabry病一种可靠且低创性的诊断方法已开始受到国内学者的关注。鉴于检验条件、患者经费不足、患者个人意愿等客观原因,我科患者未能进行α-Gal A酶活性检测和基因检测,期望在后续诊治中能够广泛开展这一检测。
Fabry病的特异性治疗为酶替代疗法(enzyme-replacement therapy,ERT),即补充缺乏的α-GLA,是目前针对Fabry病因的唯一可用的治疗方法[18,27]。近十年来,欧洲和美国已批准基因重组人α-Gal A酶产品Agalsidase α和Agalsidase β用于临床酶替代疗法,适时开始治疗可减少患者细胞内Gb3的沉积。2018年8月10日,Migalastat(Galafold)获美国药监局批准用于成年Fabry病的治疗,是首个Fabry病口服治疗药,也是近15年来首个在美国获批的Fabry病新型治疗方法。与酶替代治疗不同的是,Galafold可增加机体内酶活性,为患者带来了福音。除美国外,Migalastat在澳大利亚,加拿大、以色列、日本、韩国和瑞士等国家也已获批,目前在中国也获批,并已开展相关临床工作。
重组人α-Gal A产品Agalsidase α和Agalsidase β以及Migalastat等新药的问世,给Fabry病患者的治疗带来历史性突破,使Fabry病患者正常存活变成现实。但目前α-Gal A酶替代疗法应用时间尚短,治疗病例数有限,对长期α-Gal A酶替代治疗的疗效、安全性等还有待进一步研究[28]。我科患者均采取了针对CKD的综合治疗,包括限制盐摄入、戒烟、降压、调脂治疗,优质低蛋白饮食,并使用血管紧张素转化酶抑制剂或血管紧张素受体拮抗剂保护残余肾功能,降低尿蛋白的排泄,以及避免使用肾毒性药物等,定期随访患者肾功能尚稳定。
猜你喜欢
上海人大月刊(2022年4期)2022-04-14
昆明医科大学学报(2021年12期)2021-12-30
中老年保健(2021年4期)2021-08-22
东方考古(2021年0期)2021-07-22
摄影之友(影像视觉)(2020年2期)2021-01-14
中国卫生(2016年1期)2016-11-12
中国卫生标准管理(2015年17期)2016-01-20
中国继续医学教育(2015年2期)2016-01-06
中外医疗(2015年11期)2016-01-04
医学美学美容·中旬刊(2015年2期)2015-10-21
