曲贝替定的合成研究进展
2020-10-16 05:41李火明张富尧
高等学校化学学报 2020年10期
李火明,张富尧
(上海时莱生物技术有限公司,上海 201210)
曲贝替定(1,ET-743)是从加勒比海鞘中分离获得的一种四氢异喹啉类生物碱[1](Scheme 1).由于曲贝替定对肿瘤细胞的体外抑制效果及对啮齿类动物和人类肿瘤模型的体内抑制效果均非常显著[2],因此快速引起了制药公司的注意.1994年,西班牙制药公司Pharma Mar获得了曲贝替定的化合物专利权,并将其推进临床.作为首个来源于海洋的抗肿瘤药物,2007年,曲贝替定被欧盟批准用于进展期软组织肉瘤的二线治疗.2015年,美国食品及药物管理局(FDA)批准曲贝替定用于治疗不可切除的或转移性脂肪肉瘤或平滑肌肉瘤的二线治疗.目前,曲贝替定的适应症已经扩展到用于治疗铂类药物敏感的复发性卵巢癌.曲贝替定的化合物专利申请于1987年[3],至今已过期,国内已有多家企业立项对曲贝替定进行仿制.曲贝替定结构复杂,具有多个连续的手性中心,仿制难度大,基于曲贝替定结构的特殊性和合成难度,许多研究者一直致力于通过有机合成方法学开发新的合成策略.

Scheme 1 Retrosynthetic analysis of trabectedin
曲贝替定是一个多环化合物,共有9个环,其中AB,DE和GH环分别组成了3个四氢异喹啉结构,F环为十元含硫桥环,再加上7个手性中心,造成了曲贝替定结构的独特性和复杂性.按照逆合成路线的分析,曲贝替定的合成策略均为最后引入GH四氢异喹啉环.根据十元环成环时官能团活化位点的不同,可以获得2种不同的双四氢异喹啉化合物(Scheme 1).因此,曲贝替定的合成策略差异主要体现在对2种双四氢异喹啉结构的构筑上.由于A,E和G环为苯环,一般直接来源于商品化的原料,本文综述了自1996年Corey等[4]首次报道曲贝替定全合成以来至今的全合成、半合成和形式合成路线.概述具有较大共通性的H和F环的合成,重点讨论B,C和D环的合成理念及其中包含的手性中心的构建思路,旨在为药物仿制的工艺路线选择和优化提供文献基础.
1 H环的合成
Corey等[4]利用转胺化反应,通过4-醛基吡啶盐将α-氨基酸酯2a转化为α-酮酸酯5.以3-羟基-4-甲氧基苯丙胺为原料,通过Pictet-Spengler反应,引入GH四氢异喹啉环,得到化合物6.通过三氟乙酸水解C18的甲氧基甲基醚(MOM)保护基后,在硝酸银的作用下,将化合物7中C21位的氰基转化为羟基,完成了曲贝替定的首次全合成(Scheme 2).

Scheme 2 First total synthesis of trabectedin by Corey group
在此基础上,Manzanares等[5]、Fukuyama等[6,7]、Zhu等[8]和Ma等[9]避免了使用C18酚羟基保护基,亦获得了曲贝替定的最终产物,依次完成了曲贝替定的全合成.
在Pictet-Spengler反应中,若使用5-甲氧基色胺为原料,对酮酸酯8进行环化后通过硝酸银进行氰基水解,可以获得芦比替定[10](Scheme 3).2018年8月,芦比替定被美国FDA授予治疗小细胞肺癌的孤儿药资格,于2020年2月获得上市申请优先审批权,2020年6月,FDA加速批准了芦比替定用于转移小细胞肺癌铂类药物进展后的二线治疗.
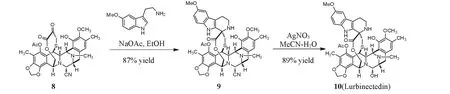
Scheme 3 Total synthesis of lurbinectedin
2 F环的合成
Corey等[4]、Manzanares等[5]、Ma等[9]和Chen等[11]以化合物3为原料,通过(PhSeO)2O氧化A环引入C10位的羟基.首先,以保护的半胱氨酸对化合物11的C22位伯醇进行选择性酯化,再经过涉及5个化学键的断裂和形成的高度串联反应,对C10位的羟基进行活化和消除得到邻亚甲基苯醌化合物14.随后通过胍试剂水解巯基保护基,并对邻甲基苯醌官能团进行Michael加成,得到化合物15,通过原位进行乙酰基保护获得化合物16.最后,根据C18和氨基上的保护基类型,可选择不同条件依次水解或同一条件同时水解2个保护基,得到化合物2b(Scheme 4).
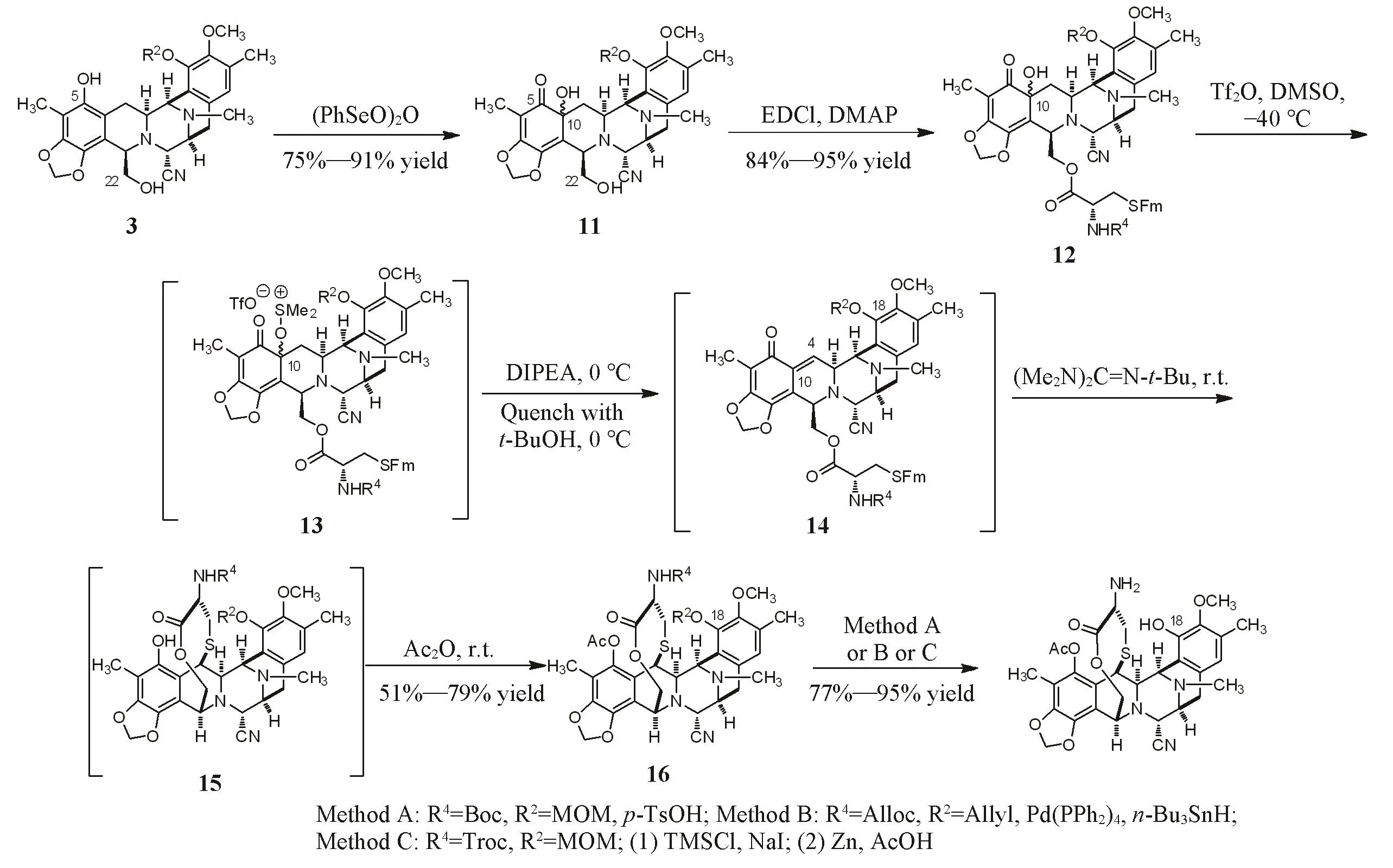
Scheme 4 F ring formation by activation at C10
Fukuyama等[6,7]和Zhu等[8]采用了不同的合成策略,如Scheme 5所示,以C4位的羟基化底物4为中间体,通过C22位的伯醇与半胱氨酸衍生物发生酯化反应得到化合物17.根据巯基上的保护基类型,可选择在碱性或酸性条件下水解保护基,随后进行分子内Michael加成环化反应,并原位用醋酸酐保护C5酚羟基.环化反应的中间体是邻亚甲基苯醌19.最后根据C18和氨基上的保护基类型,可选择不同条件依次水解或同一条件下同时水解化合物20的2个保护基,得到化合物2b.

Scheme 5 F ring formation by activation at C4
3 双四氢异喹啉结构的合成
与GH环和F环的合成策略具有的共性不同,由ABCDE环组成的双四氢异喹啉结构(化合物3和4,Scheme 1)的合成更体现了曲贝替定合成的差异性和多样性.A环和E环为苯环,通常来自市售原料.因此,BCD并三环的构建展示了不同研究组的合成理念和对有机分子的操控思路.依合成策略不同,BCD所组成的三环结构中含有5或6个手性中心.在大部分合成策略中,其中的1或2个手性中心来源于天然氨基酸的手性或通过不对称催化或手性辅基诱导产生,其余的手性中心则在反应过程中由这1~2个手性和环的构象来进行诱导.
3.1 BCD环的合成策略
Corey等[4]通过Pictet-Spengler反应构建B环,再通过第2次Pictet-Spengler反应同时构建CD环.通过逆合成推演的2个片段,即化合物21和22通过Strecker反应进行偶联.C3和C13手性中心通过双键的不对称催化氢化反应获得.C1,C11和C21手性中心通过诱导产生(Scheme 6).
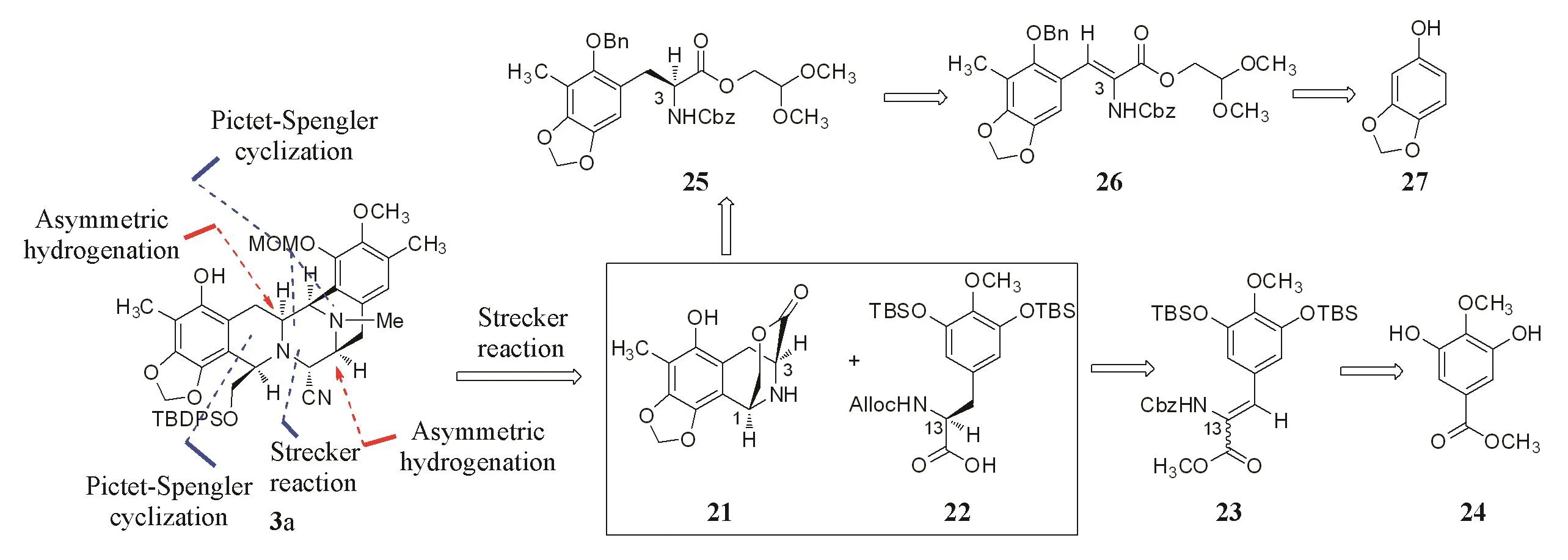
Scheme 6 Retrosynthetic analysis of BCD ring by Corey group
如Scheme 7所示,化合物21和22在醋酸和氰化钾的作用下发生Strecker反应生成α-胺基腈,建立C21手性中心,随后,将C5酚羟基保护为烯丙基醚28.将化合物28中的桥环内酯结构还原为环状半缩醛,通过氟化钾水解C16和C18位的酚羟基硅醚保护基,再在甲烷磺酸的作用下进行第2次Pictet-Spengler反应,同时构建CD环,并建立C11手性中心,得到化合物29.依次以三氟甲磺酸酯对C16位的酚羟基,叔丁基二苯基硅醚对C22位的伯醇和MOM对C18位的酚羟基进行选择性保护,得到全保护的双四氢异喹啉化合物30.在二氯二(三苯基膦)钯和三丁基锡氢的作用下,同时水解C5位的烯丙基和N12位的Alloc保护基,并通过Stille偶联将C16位的三氟甲磺酸酯基转化为甲基,得到用于合成F环的前体化合物3a.
2002年,Corey等[12]又对上述合成路线进行微调,开发了一条更加高效和更易于操作的合成工艺.化合物21与氨基酸31进行酰胺偶联和C5位酚羟基保护,得到化合物32.在二乙氧基氢化铝锂和低温(-78℃)条件下,选择性还原化合物32中C11位的酯羰基得到桥环半缩醛33.在氟化钾条件下同时水解C16和C18位的酚羟基硅保护基后,在三氟甲磺酸的作用下进行Pictet-Spengler反应,得到双四氢异喹啉骨架34.在二乙氧基氢化铝锂和更高温度(0℃)下,还原C21位的酰胺羰基,再在乙酸和氰化钾的作用下反应得到胺基腈29(Scheme 8).

Scheme 8 Optimization of the construction of BCD ring by Corey group
从海鞘中提取曲贝替定的效率极低,1000 kg海鞘中只能提取1 g曲贝替定,不能满足临床药物开发对候选化合物的用量需求.虽然Corey研究组开发了曲贝替定的全合成路线,然而该路线直线步骤包括36步,总收率仅为0.72%,也不能满足药物的长期供应.为此,Manzanares等[5]在Corey全合成路线的基础上,开发了以Cyanosafracin B(35)为原料的半合成路线.Cyanosafracin B[13]是一种生物发酵的产物,可以进行kg级的规模生产.且Cyanosafracin B具有与曲贝替定完全相同的五环骨架立体构型,其余结构也高度相似.因此,只需要对官能团进行适当的转化和修饰便可以得到双四氢异喹啉关键中间体3b.对Cyanosafracin B的伯胺和C18位酚羟基分别进行叔丁氧羰基(Boc)保护和MOM保护,通过氢氧化钠水解C7位甲氧基得到化合物36.在Pd/C条件下对苯醌36进行还原,得到苯三酚.用ClCH2Br保护C7/C8位邻苯二酚官能团,并对C5位酚羟基进行烯丙基醚保护得到化合物37.将胺基的Boc保护基水解后,通过Edman降解水解肽键,脱除丙氨酸片段,得到化合物38.为了保护C18位酚羟基,需要对C22位胺基先后进行三氯乙氧基羰基(Troc)保护和水解.最后,采用亚硝酸钠和乙酸将C22位胺基转化为羟基,得到用于合成F环的前体化合物3b(Scheme 9).最终合成曲贝替定的总步骤减为21步,总收率约1%.该路线也成为Pharma Mar公司对曲贝替定的商业化合成路线.
2002年,Fukuyama等[6]完成了曲贝替定的全合成,在双四氢异喹啉中间体的合成上,采取了与Corey完全不同的策略.通过乌吉(Ugi)四组分反应实现对2个复杂程度相近的手性胺片段41和手性酸片段42的偶联.其中手性片段41的C1手性中心通过他们发展的手性辅基诱导不对称Mannich反应构建,片段42的C13手性通过不对称氢化反应构建[14].依次采用内酰胺化反应形成C环,Heck反应形成D环,傅克反应形成B环(Scheme 10).
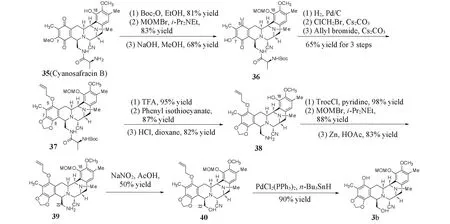
Scheme 9 Semi-synthesis of BCD ring by Pharma Mar group

Scheme 10 Retrosynthetic analysis of BCD ring by Fukuyama group
Ugi四组分反应高收率地偶联手性胺41和手性酸42,形成一对非对映异构体.将化合物50的C22位羟基保护基转换为乙酰基,在三氟乙酸条件下脱除N12位胺基Boc保护基后,所得产物在乙酸乙酯中直接加热进行分子内酰胺化,形成内酰胺51,完成C环的构建.依次保护化合物51的C5位酚羟基和N12位胺基,再对内酰胺官能团进行还原和脱水形成烯烃52,同时也消除了因Ugi反应非对映选择性差对后续反应的影响.通过化合物52的分子内Heck环化反应成功构建了D环,并形成了C11手性中心.将化合物53的C5位酚羟基保护基转化为乙酰基,并将N12位胺基保护基转化为Troc,采用二甲基过氧化酮对环外双键进行环氧化后,以甲醇进行环氧开环得到化合物54.氰基硼氢化钠还原引入C3手性中心.将化合物55的C4位伯醇保护为叔丁基二甲基硅醚,水解C5和C22位的2个乙酰基保护基后,将C5位酚羟基选择性保护为苄醚,并用红铝还原内酰胺,原位环化得到噁唑烷化合物56.三甲基硅氰(TMSCN)对噁唑烷56进行开环,引入α-胺基腈结构,构建了C21手性.随后,乙酰基保护C22位羟基,水解C4位羟基保护基,再通过戴斯-马丁氧化剂氧化得到醛57.钯炭氢化还原C5苄基保护基时,原位发生傅克反应得到化合物58,在构建B环的同时确立了C4的手性中心.将化合物58的C18位酚羟基选择性保护为烯丙基醚,水解C22位乙酰基保护基,得到用于F环合成的关键中间体4a(Scheme 11).该合成路线首次采用C4位羟基作为活化基团进行十元含硫桥环的合成,并被多个研究组利用和借鉴,作为曲贝替定形式合成的桥联关键中间体.该方法巧妙地使用Ugi四组分缩合和Heck分子内环化反应合成双四氢异喹啉骨架,但是合成过程中多次使用和更换保护基,导致部分操作繁琐,总的合成路线达到50步,总收率为0.56%.
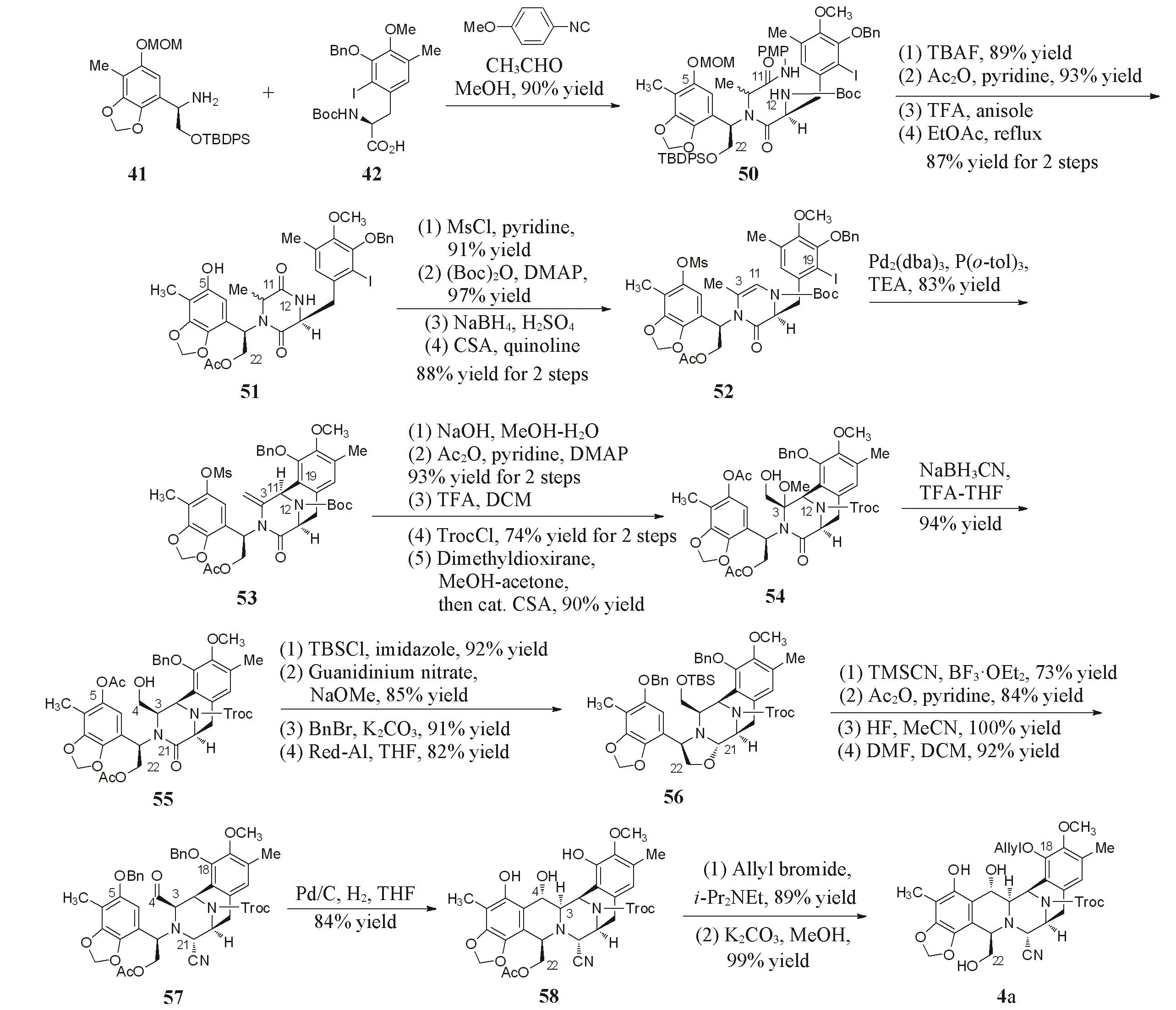
Scheme 11 Construction of BCD ring by Fukuyama group
2006年,Zhu等[8]完成了曲贝替定的全合成.在双四氢异喹啉结构的合成上,依次通过Pictet-Spengler反应构筑D环,Strecker反应构筑C环,Friedel-Crafts反应构筑B环.通过手性胺基片段59对消旋的溴代物片段60进行N-烷基化拼接2个片段.最先引入的C3手性来自于丝氨酸,含C3手性的Garner醛62发生Pictet-Spengler反应,以单一的非对映异构体同时构建了B环和C11手性中心,得到片段59.通过Corey等开发的手性相转移催化剂介导的不对称烷基化形成C13手性[15](Scheme 12).手性相转移催化技术是合成复杂手性氨基酸的重要途径,我们[16]也采用该手性相转移催化剂,以化合物67为原料合成化合物68,进行了C3手性中心的构建,对映体过量值(e.e.)可达95%(Scheme 13).该方法目前已经放大到kg级的规模,用于曲贝替定原料的大量供应.
在碱性条件下,手性胺59与消旋的溴代物60通过SN1机理发生N-烷基化反应,以3∶1的非对映体比例(d.r.)形成C1手性.依次保护化合物69的C4位伯醇,水解C13位乙酰基,得到化合物70.将化合物70的C21位醇羟基氧化为醛后,在TMSCN的作用下发生Strecker反应得到胺基腈71,同时构建了C环和C21手性中心.硼氢化锂还原化合物71的C22位酯基,用乙酸酐保护所得的伯醇,氢氟酸水解C4位硅保护基后,用戴斯-马丁氧化剂氧化C4位醇羟基得到醛72.三氟乙酸脱除C5位酚羟基MOM保护基时,与C4位醛基原位发生傅克反应形成B环和C4手性中心.最后通过碳酸钾水解C22位羟基保护基得到用于构建F环的关键中间体4b(Scheme 14).合成曲贝替定的总步骤为31步,总收率为1.7%.
Danishefsky等[17]采用Friedel-Crafts反应构建B环,并利用Pictet-Spengler反应同时形成C环和D环.胺片段73和酸片段74[18]通过酰胺化反应偶联.2个手性片段的手性中心C1和C13分别由酮羰基的不对称加氢还原和双键的不对称氢化产生(Scheme 15).

Scheme 12 Retrosynthetic analysis of BCD ring by Zhu group
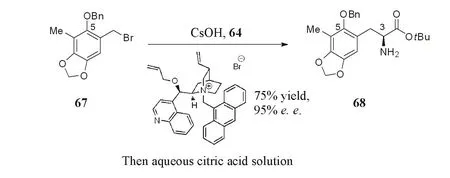
Scheme 13 Construction of C3 stereo center by chiral phase transfer catalyst

Scheme 14 Construction of BCD ring by Zhu group

Scheme 15 Retrosynthetic analysis of BCD ring by Danishefsky group
手性胺片段73和手性氨基酸片段74进行酰胺偶联后,以二氯二氰基苯醌(DDQ)脱除化合物80的C11位对甲氧基苄基(PMB)保护基,在Cu(OTf)2条件下对C4位仲醇进行脱水形成烯烃,氧化C11位伯醇,并水解C5位烯丙基保护基,得到化合物81.通过筛选酸试剂,发现在二氟乙酸条件下,N12位胺基Boc保护基发生水解,同时进行Pictet-Spengler反应,一步构建了C环、D环和C11手性中心,反应收率为42%~58%.对化合物82的C5位酚羟基进行叔丁基二甲基硅醚保护,并将N12位甲基转化为Troc保护基,得到化合物83.通过2步反应将C5位硅保护基更换为MOM基,得到化合物84.以二甲基过氧化酮(DMDO)对化合物84的C3/C4位双键进行环氧化,形成C4手性中心,原位加入大量的氰基硼氢化钠进行还原,确立C3手性,得到化合物86.Pd/C还原脱除2个苄基保护基后,以二异丁基氢化铝(Dibal-H)和丁基锂的复合试剂对内酰胺进行半还原,形成噁唑烷87.将化合物87的C18位酚羟基保护为烯丙基醚,再用氰化钾和乙酸对噁唑烷进行开环,获得C21手性中心,最后在三氟乙酸条件下对C5位MOM保护基进行水解,得到了Fukuyama路线[6]的中间体4a(Scheme 16),完成了曲贝替定的形式合成.

Scheme 16 Construction of BCD ring by Danishefsky group
Williams等[19]通过自由基环化形成B环,同时发生Pictet-Spengler反应构建CD双环.然而,在Pictet-Spengler环化时,反应的区域选择性仅为0.72∶1,其中,次要异构体为所需的构型.2个片段通过胺和酰氯的酰胺化反应进行偶联,其中片段89中的C13手性通过不对称烷基化反应产生[20],而片段88中的C3手性来自于丝氨酸.其它手性则在反应过程中由这2处手性和环构型诱导产生.在后续的C4位羟基构建中借鉴了Danishefsky等[17]策略中的环氧化和还原组合,并沿用了Fukuyama等[6]发现的中间体86,完成曲贝替定的形式合成(Scheme 17).
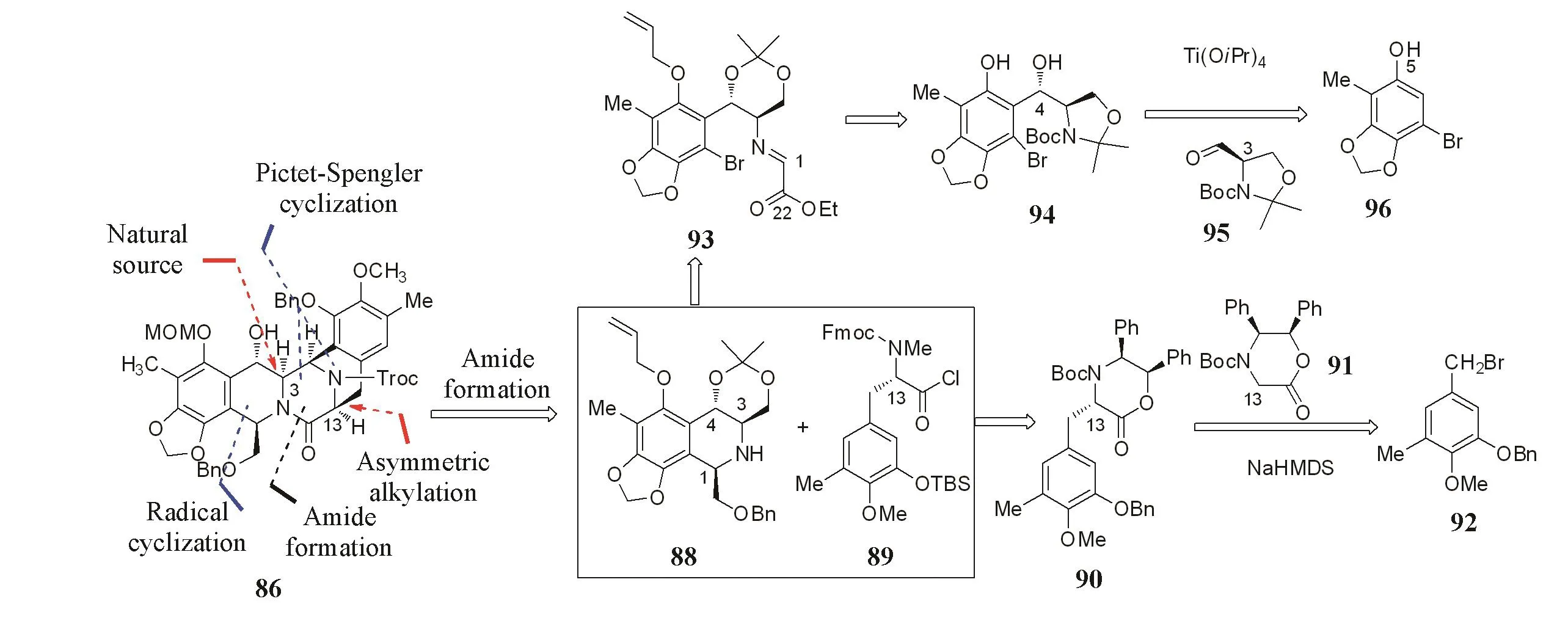
Scheme 17 Retrosynthetic analysis of BCD ring by Williams group
碱性条件下酰氯89和胺基醇88进行酰胺化反应,并将N12位胺基保护基由芴甲氧羰基(Fmoc)更换为Boc,得到化合物97.酸性树脂水解化合物97的丙酮叉保护基的同时,处于苄位的C4位活性醇羟基被甲醇取代,发生C4手性中心的差向异构化,得到1组等量的非对映异构体.四丁基氟化铵(TBAF)水解叔丁基二甲基硅醚保护基,对C11位伯醇进行Swern氧化得到醛和半胺缩醛的混合物98.在三氟乙酸条件下,对化合物98进行Pictet-Spengler环化,同时形成了CD环和C11手性中心,以72%的总收率得到环化产物,但是反应的区域选择性仅为0.72∶1(99a∶99b).将化合物99a的N12位胺基转化为Troc保护基,保护C18位酚羟基为苄醚,在四三苯基膦的作用下水解C5位烯丙基保护基,并重新保护为MOM基,得到化合物84.化合物84为Danishefsky等[17]提出路线的中间体,因此Williams等沿用Danishefsky等[17]的合成方案,将化合物84衍生为Fukuyama等[6]合成路线的中间体化合物86,完成曲贝替定的形式合成(Scheme 18).

Scheme 18 Construction of BCD ring by Williams group
由于第一代反应路线长达50步,总收率为0.56%,Fukuyama等[7]开发了第二代合成路线.该路线采用完全不同的策略来合成双四氢异喹啉结构.C3的手性中心来源于谷氨酸103,随后依次通过C3手性和环构型的诱导产生其它5个手性中心.通过Pictet-Spengler反应同时构建CD环,傅克反应合成B环.2个片段的偶联依赖重氮盐101的Heck反应进行(Scheme 19).在新的路线中,经过28步完成曲贝替定的全合成,总收率为1.1%.

Scheme 19 Retrosynthetic analysis of BCD ring by Fukuyama group
谷氨酸103衍生为化合物107后,其双键通过底物诱导进行非对映选择性氢化获得C13手性,在水合肼的条件下水解胺基乙酰基保护基,并用硼氢化钠选择性还原非保护的内酰胺得到中间体108.后者在三氟乙酸条件下进行Pictet-Spengler环化反应,同时形成CD环和C11手性.化合物109经过4步衍生形成烯烃片段102(Scheme 20).
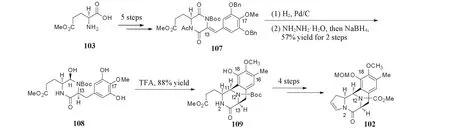
Scheme 20 Synthesis of cyclic olefin 102
通过亚硝酸叔丁酯在三氟化硼乙醚作用下将芳基胺104转化为重氮盐101后,与烯烃片段102进行Heck反应,引入C1手性.通过四氧化锇和铁氰化钾将化合物110的C4/C22双键进行双羟化得到化合物111后,采用高碘酸氧化形成二醛112,再水合后形成环状半缩醛113.氢化还原脱除化合物113的C5位苄基保护基,所得化合物114在热的二甲苯溶液中发生傅克反应,构建B环和C4手性中心.化合物115的醛和内酰胺官能团在红铝的作用下进行还原和环化,得到噁唑烷化合物116.在氰化钾和乙酸的条件下对化合物116进行开环反应得到胺基腈,形成C21手性中心,得到用于F环合成的关键双四氢异喹啉中间体4c(Scheme 21).
Ma等[9]采用Pictet-Spengler环化反应合成B环,通过另一次Pictet-Spengler环化反应合成D环,并利用Strecker反应构建C环.2个片段通过Pictet-Spengler反应进行偶联.他们精确分析了2个片段结构的相似性,以提供C3手性的酪氨酸的氨基醇片段63为原料,结合光催化碳氢键活化反应,合成了第2个片段117,大大缩短了反应的总操作(Scheme 22).最终以26步的总步骤,1.6%的总收率合成了曲贝替定.

Scheme 21 Construction of BCD ring by Fukuyama group
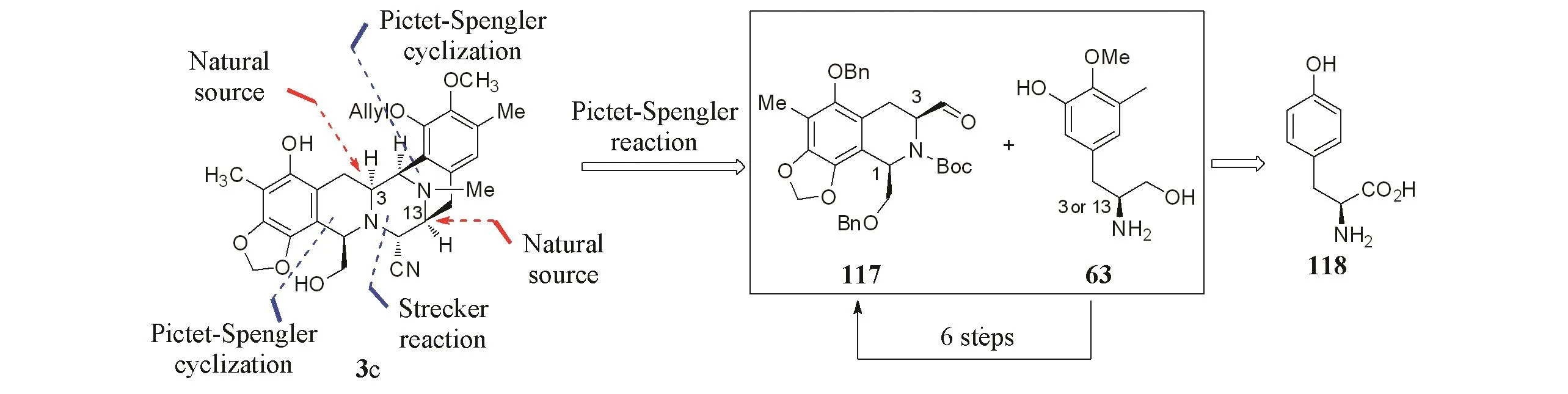
Scheme 22 Retrosynthetic analysis of BCD ring by Ma group
化合物63的合成借鉴Chen等[21]的路线,以酪氨酸118为原料经过8步反应合成.化合物63既作为后续Pictet-Spengler反应的一个片段,也作为合成另一个片段117的中间体.苄氧基乙醛与化合物63在乙酸条件下进行Pictet-Spengler反应,并对胺基进行Boc保护,得到化合物119.钴催化氧化苯酚119得到苯醌120,利用光催化进行C7位甲氧基的碳氢键活化反应,形成苯并二氧杂环戊烯(Benzo[1,3]dioxole)[22],经过优化,在450 nm的蓝光条件下反应,收率高达84%,且反应已经放大到数十克的规模而不影响收率和选择性.保护化合物121的C5位酚羟基后,Swern氧化C11位伯醇得到醛117(Scheme 23).

Scheme 23 Synthesis of chiral amine 63 and chiral aldehyde 117
在乙酸条件下,化合物117和化合物63进行Pictet-Spengler环化反应,构建D环和C11手性,得到化合物122.对化合物122的N12位胺基进行还原甲基化.随后,选择性保护C18位的酚羟基得到化合物123.将C21位伯醇进行Swern氧化后,采用三氟乙酸脱除N2位胺基Boc保护基,在TMSCN条件下进行Strecker反应,构建C环和C21手性中心,以大于10 g的规模得到化合物124.在三氯化硼条件下水解化合物124的苄基的同时发生了少量的氰基水解,最后,用TMSCN将水解产物完全转化为胺基腈3c(Scheme 24).后续反应按照Corey等[4]提出的路线得到1.1 g曲贝替定.
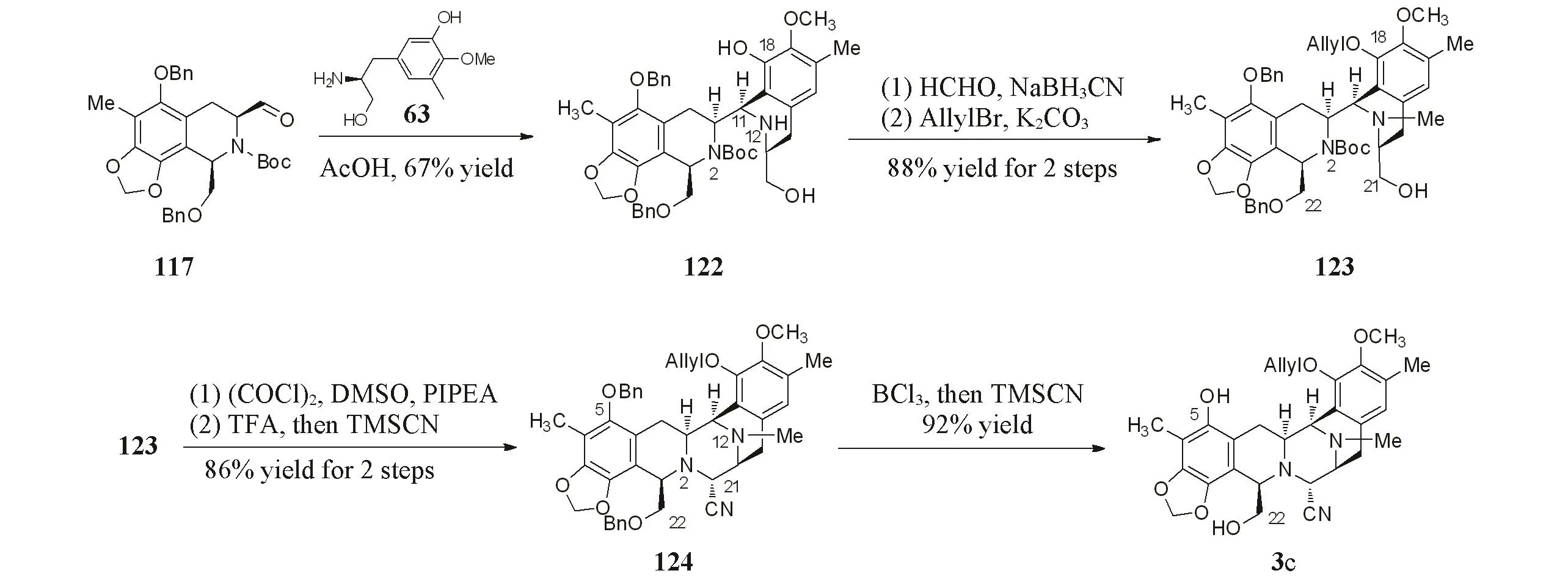
Scheme 24 Construction of BCD ring by Ma group
Chen等[11]采用Bischler-Napieralski反应构建B环,Pictet-Spengler反应构建D环及Strecker反应构建C环.片段125和63通过Pictet-Spengler反应拼接.片段63的C13手性来自于酪氨酸,片段125的C3手性来源于丝氨酸(Scheme 25).
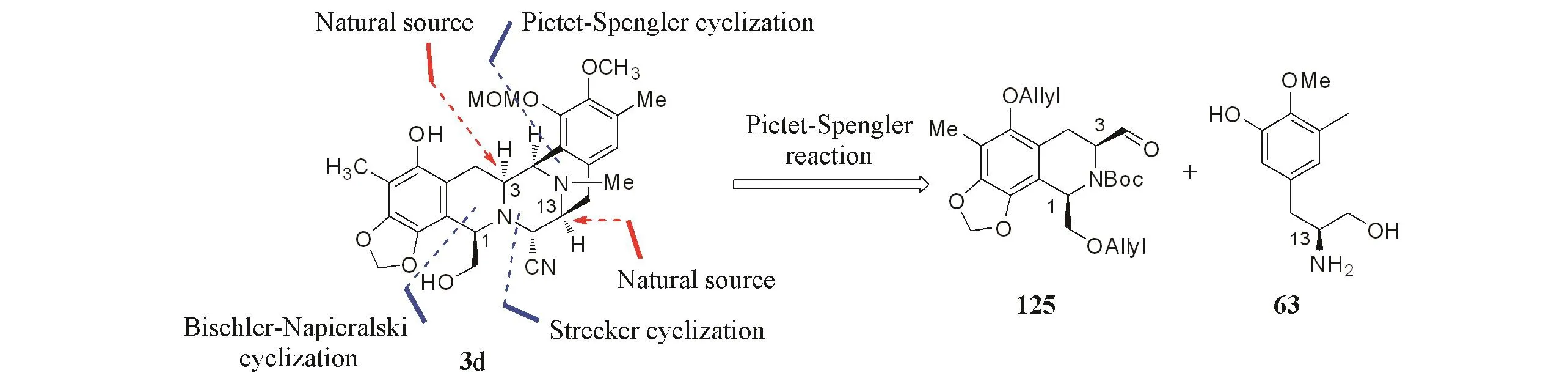
Scheme 25 Retrosynthetic analysis of BCD ring by Chen group
芝麻酚27经3步转化为甲基芝麻酚106后,与来源于丝氨酸的Garner醛95在甲基氯化镁的作用下进行加成得到化合物126[23].经还原消除苄位碳氧键和保护基转化得化合物127.后者在三氯氧磷的条件下关环得到二氢异喹啉,形成B环,并用硼氢化钠还原产生C1手性中心,得到四氢异喹啉化合物128,其中C1手性的非对映选择比例为6∶1.对化合物128进行胺基Boc保护,再氧化C11位伯醇得醛125(Scheme 26).
醛125与氨基醇63在乙酸和亚磷酸二苄酯的作用下进行Pictet-Spengler反应形成D环.依次对化合物129的N12位胺基进行烯丙基氧基羰基(Alloc)保护,C18位酚羟基进行烯丙基保护,盐酸甲醇水解N2位胺基Boc保护基,得到化合物130.对化合物130的C22位醇羟基进行Swern氧化,并在TMSCN和ZnCl2条件下进行Strecker反应,构建C环和C21手性中心,得到双四氢异喹啉骨架化合物131.最后,通过叔丁基二甲基氯硅烷(TBSCl)选择性保护C22位醇羟基和C5位酚羟基,对裸露的C18位酚羟基进行MOM保护,再水解C22和C5位叔丁基二甲基硅醚保护基,得到用于合成F环的关键中间体3d(Scheme 27).

Scheme 26 Synthesis of aldehyde fragment 125
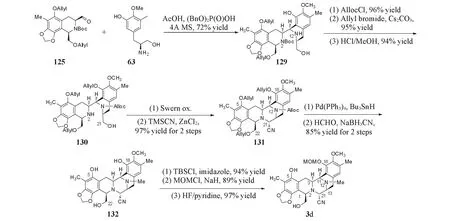
Scheme 27 Construction of BCD ring by Chen group
3.2 BCD环的合成策略小结
最经典的合成四氢异喹啉结构的方法依然是Pictet-Spengler环化反应[24].该方法不仅能温和地形成所需的环状结构,还能有效地通过成环过程中的过渡态构象高效地控制新产生的手性中心.通过精心设计成环位点及官能团,能够同时构建2个环骨架.傅克反应与Pictet-Spengler反应相似,也是利用富电子苯环的亲电加成来进行环化.这两种反应类型在曲贝替定的环合成中使用率达到65%,并主要用于形成B环和D环.C环中存在的α-胺基腈官能团主要通过Strecker反应[25]产生,因此Strecker反应也常用于C环的合成.此外,Heck反应和自由基反应也被用于一些特殊设计的底物环化.为了提高全合成的效率,曲贝替定通常采用汇聚式合成,即分别合成2个复杂程度相当的片段,再通过偶联反应进行组装.在偶联反应类型的选取上,酰胺化反应属于传统而经典的操作,Pictet-Spengler反应、Ugi反应和Heck反应因高效和高选择性也被用于片段偶联策略.双四氢异喹啉骨架中虽然存在5或6个手性中心,但是由于结构中的多环骨架,大部分手性中心的建立只需要依赖于预先组装的1或2个手性中心,并在反应过程中通过位阻或过渡态的构象来进行控制.对于该预先设置的1或2个手性中心,大部分课题组选择优先建立C3和C13手性,少数课题组选择建立C1手性,再诱导产生其它手性中心.手性氨基酸原始手性保持,不对称氢化以及手性辅基诱导的不对称合成均可作为该1或2个手性的来源(表1).

Table 1 Summary of synthetic strategies for construction of BCD ring
4 总结与展望
自曲贝替定被发现以来,到目前为止已有5例全合成,1例半合成和3例形式合成报道.Corey等[4]对GH环合成的先驱工作为其他课题组的合成奠定了良好的基础.Corey等[4]和Fukuyama等[6]分别开发了十元含硫桥环F环的合成策略,也分别被后续的研究组借鉴.因此,ABCDE五环所组成的双四氢异喹啉结构,特别是BCD并三环的合成为各研究组提供合成理念.Manzanares等[5]的半合成策略以生物发酵产物Cyanosafracin B为原料,本身已经具备曲贝替定双四氢异喹啉骨架的五环结构和所有手性中心,因此仅需要进行适当的官能团转换和保护基变换.除目前正在商业化的半合成路线外,曲贝替定的合成步骤从26~50步不等.串联反应是最有效的提高合成效率的方式,通过官能团设计,同时组建多个化学键.而保护基的频繁更换使合成路线显得繁琐,每更换1次保护基至少需要3步反应.因此,高度串联、高选择性的有机合成方法学日益受到重视[26].避免保护基的使用也成为天然产物全合成中的一种趋势[27].此外,精确分析骨架的对称性[28],使用相同的高级中间体合成不同的片段,能够缩短合成路线的步骤,极大地提高全合成和药物合成的效率.通过不断优化曲贝替定的合成路线,将逐渐降低药品生产的成本,造福全世界的患者.
猜你喜欢
分子催化(2022年1期)2022-11-02
色谱(2020年3期)2020-02-12
火炸药学报(2019年5期)2019-11-11
天然气与石油(2018年6期)2019-01-29
中成药(2017年7期)2017-11-22
国外医药(抗生素分册)(2016年4期)2016-07-12
国外医药(抗生素分册)(2016年2期)2016-07-12
国外医药(抗生素分册)(2016年1期)2016-07-10
郑州大学学报(理学版)(2014年3期)2014-03-01
原子与分子物理学报(2014年4期)2014-02-28