
采用宏基因组学分析肺结核患者与健康人群呼吸道微生物菌群的特征
2020-10-13 07:51邱倩刘东鑫刘春法邱捷严晓峰赵雁林
中国防痨杂志 2020年10期
邱倩 刘东鑫 刘春法 邱捷 严晓峰 赵雁林
结核病目前仍是全球前10位死因之一,同时自2007年以来一直位居单一传染性疾病死因之首。中国是全球30个高负担国家之一,如果没有充足的防治和研究资金投入,以及新的诊断工具和预防结核感染的新疫苗、新药品出现,联合国可持续发展目标和终止结核病策略目标将难以实现[1]。目前经典的结核病诊断方法主要集中在结核分枝杆菌的识别上,然而,结核病的病程进展不仅仅体现在疾病的病原体本身上,还由局部微生物菌群和免疫因子之间相互作用决定。
微生物菌群是一组病原微生物集落的总称,广泛存在于人体体表及与外界相连的腔道上,如口腔、呼吸道、肠道、泌尿生殖道等部位。既往认为正常下呼吸道系统是无菌的,而近年研究证实呼吸道广泛存在一定种类和数量的微生物,尤其集中于下呼吸道的黏膜层及上皮表面,起到维持气道的正常结构和功能,抵御、拮抗外袭菌群和产生生理活性物质调节免疫的作用[2]。越来越多的研究认为疾病的状态、抗生素治疗、环境因素、饮食结构、社会人口学因素等可影响微生物菌群的类型和分布;比如,研究认为肺结核患者和健康人群的呼吸道菌群的组成就存在差异[3-4]。而新的核酸测序技术在肺结核患者呼吸道菌群多样性特征上的应用,也为探索结核病的病理机制提供了新的方向。然而,在有限的相关研究中,不同研究发现结核病特征菌群的种类不一致,结核病相关菌群的丰度和多样性也有很大差异。因此,笔者综合回顾了不同地区、不同样本关于呼吸道菌群与肺结核关系的研究,进行系统评价及宏基因组学分析,以期了解肺结核和呼吸道菌群的确切关系,为肺结核及其并发肺部感染的发病机制及未来的治疗策略提供新的理论依据。
资料和方法
一、检索策略
检索所有包含结核和呼吸道菌群的中文和英文文献。计算机检索英文数据库PubMed、Embase and Ovid、Web of Science、Google Scholar、Cochrane Library和中文数据库中国知网、万方数据、中国生物医学文献服务系统与维普网,时间限定为2010年1月1日至2019年12月31日,以“结核”或“肺结核”和“呼吸道菌群”或“气道菌群”或“肺菌群”或“痰菌群”和“健康对照”为中文主题词和关键词,以“tuberculosis”或“lung tuberculosis”和“respiratory microbiota”或“airway microbiota”或“lung microbiota”或“sputum microbiota”和“healthy controls”为英文主题词和自由词进行检索。
二、文献纳入与排除标准
1.纳入标准:(1)病例组为经临床、影像学及实验室检查(细菌培养及药敏检测)确诊为肺结核患者,对照组为健康人群,研究人群为全球患者;(2)研究设计为前瞻性试验研究;(3)研究内容为呼吸道菌群;(4)研究方法为微生物的宏基因组测序;(5)研究中提供呼吸道菌群数量及分布的原始数据,各研究要求独立;(6)若同一研究小组发表2篇以上相关论文,只选取最近的包含大样本的研究论文。
2.排除标准:(1)非人类研究;(2)非呼吸道菌群的研究;(3)不能计算呼吸道菌群数量及分布的有效数据的综述和报道;(4)特殊人群,如并发糖尿病、HIV阳性或艾滋病、器官移植后长期使用免疫抑制剂的患者等相关的论文。
三、文献信息和数据提取
采用纽卡斯尔-渥太华质量评价表(newcastle-ottawa quality assessment scale,NOS)单独进行文献质量评价,其中,低质量文献排除出最终分析中。采用Excel 2013记录原始文献基本信息和数据提取表格,文献基本信息包括第一作者、文献发表年份、研究所在国家、样本人数(病例组和对照组)、测序方法、16S rRNA测序区域、元数据等内容,以及荟萃分析优先报告的条目(systematic reviews and meta-analyses,PRISMA)的各条目内容;数据提取表格包括元数据的元信息、数据列表等数据。在数据提取和质量评价之前,先对各评价量表的使用进行培训与评价,并讨论数据提取和评价过程中可能遇到的问题,对评价内容取得一致性认可后再进行数据提取。文献选择和提取过程由一名研究者单独完成,另一名研究者进行独立核对,评价纳入文献的质量及核准提取数据的准确及完整,如遇分歧则讨论解决或交由第三方决定达成一致。
四、序列构建扩增序列变体(amplicon sequence variant,ASV)构建和物种注释
从公共数据库下载测序数据和与原作者联系获得原始测序数据后,采用QIIME2软件对原始数据进行质控及预处理,统一处理参数,过滤低质量序列和嵌合体,去除重复序列。利用DADA2(ver1.8)包对筛选过的ASV表[类似传统的操作分类单元(operational taxonomic units,OTU)表,但更精确];使用朴素贝叶斯机器学习分类器方法,利用Greengene数据库对99%相似水平的ASV代表序列进行比对和物种注释,获得注释的物种信息分类,并分别在各个分类学水平统计各样本的群落组成。
五、Alpha(α)多样性分析
α多样性是对单个样本物种多样性的分析,包括观测到的OTU数目、Shannon指数、Chao1指数、系统发育多样性(Faith’s phylogenetic diversity,Faith PD)指数、Evenness指数等。由于不同研究存在不同的测序深度,故在对数据进行α多样性分析之前,对ASV表进行标准化处理。使用QIIME2软件完成α多样性分析。
六、Beta(β)多样性分析
β多样性是指样本与样本之间的多样性,反映每个样本组内或样本间的物种群落组成差异的大小。经过统一各样本的序列数后,设置统一的抽样深度,随机抽取序列,生成新的biom类型文件,计算β多样性距离(Jaccard距离、Bray-Curtis距离、加权UniFrac距离和未加权UniFrac距离),进行主坐标(principal coordinates analysis,PCoA)分析和PERMANOVA分析。
七、统计学处理
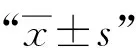
结 果
一、文献基本情况

图1 文献检索流程图
文献检索流程图(图1)显示,共检索到9327篇论文,剔除不同数据库收录的重复论文4826篇后,得到论文4501篇,阅读每篇论文的标题和摘要了解各论文的研究内容后,剔除论文共4189篇,包括2165篇综述、评论、专利和社论,386篇会议摘要,798篇基础研究,247篇非宏基因组测序技术论文,593篇非人类研究,剩余312篇论文,经阅读全文并对论文进行NOS评分,剔除303篇论文(包括235篇无相关数据,42篇重复数据,11篇操作指南,15篇采用痰普通培养方法的论文)后,最终纳入论文9篇,NOS评分均大于5,全为英文论文。
纳入的9篇文献均发表于2012—2019年之间,其中7项研究采用Roche/454测序平台进行DNA测序,2项研究分别采用Ion Torrent和Illumina平台进行测序,仅剩1项未知。研究地域方面,4项研究发生在中国,其余是哥伦比亚、美国、南非、印度和墨西哥各1项。研究标本方面,除了Vázquez-Pérez等[5]的研究的肺结核组和健康对照组均采用了支气管肺泡灌洗液(bronchoalveolar lavage fluid, BALF)样本,其余肺结核组均选择了痰标本,而健康对照组则选择了不同类型的呼吸道分泌物作为样本。其中5项研究可获得原始测序数据。纳入文献的基本信息见表1。
二、肺结核患者和健康人群呼吸道菌群群落结构组成差异
为了解两组呼吸道菌群的定性差异,我们首先运用iTOL可视化平台利用统一参数分析菌群的聚集特征。系统发育树表明,虽然来自5项不同的研究有不同的测序深度和样本大小,但肺结核患者和健康人群的高丰度菌群分类单元均有明显的聚类,而两者明显不同,提示呼吸道菌群主要按照疾病进行聚类(图2)。
两组共检测到19个门,39个纲,57个目,288个科,564个属,369个种。两组在门水平相对丰度排在前10位的共有菌群是:厚壁菌门(Firmicutes)、OD1门、变形菌门(Proteobacteria)、拟杆菌门(Bacteroidetes)、放线菌门(Actinobacteria)、梭杆菌门(Fusobacteria)、蓝菌门(Cyanobacteria);但门水平相对丰度的构成比差异较大,和健康对照组相比,肺结核组的OD1门相对丰度较高(45.54% 和0.16%),而Firmicutes(1.28%和37.84%)、Proteobacteria(0.41%和14.38%)、Bacteroidetes(0.37%和27.38%)、Actinobacteria(0.15%和10.45%)的相对丰度较低,见图3。在属水平上,肺结核组相对丰度排在前10位的属分别是OD1门下的属、Actinomycetales目下的属、Lactobacillus、Streptococcus、Acinetobacter、Prevotella、Flavobacteriaceae科下的属。健康对照组前10位的属水平物种组成依次为Prevotella、Streptococcus、Veillonella、Corynebacterium、Fusobacterium、Haemophilus、Prevotella、Clostridiales目下的属、Neisseria、Propionibacterium(图4)。
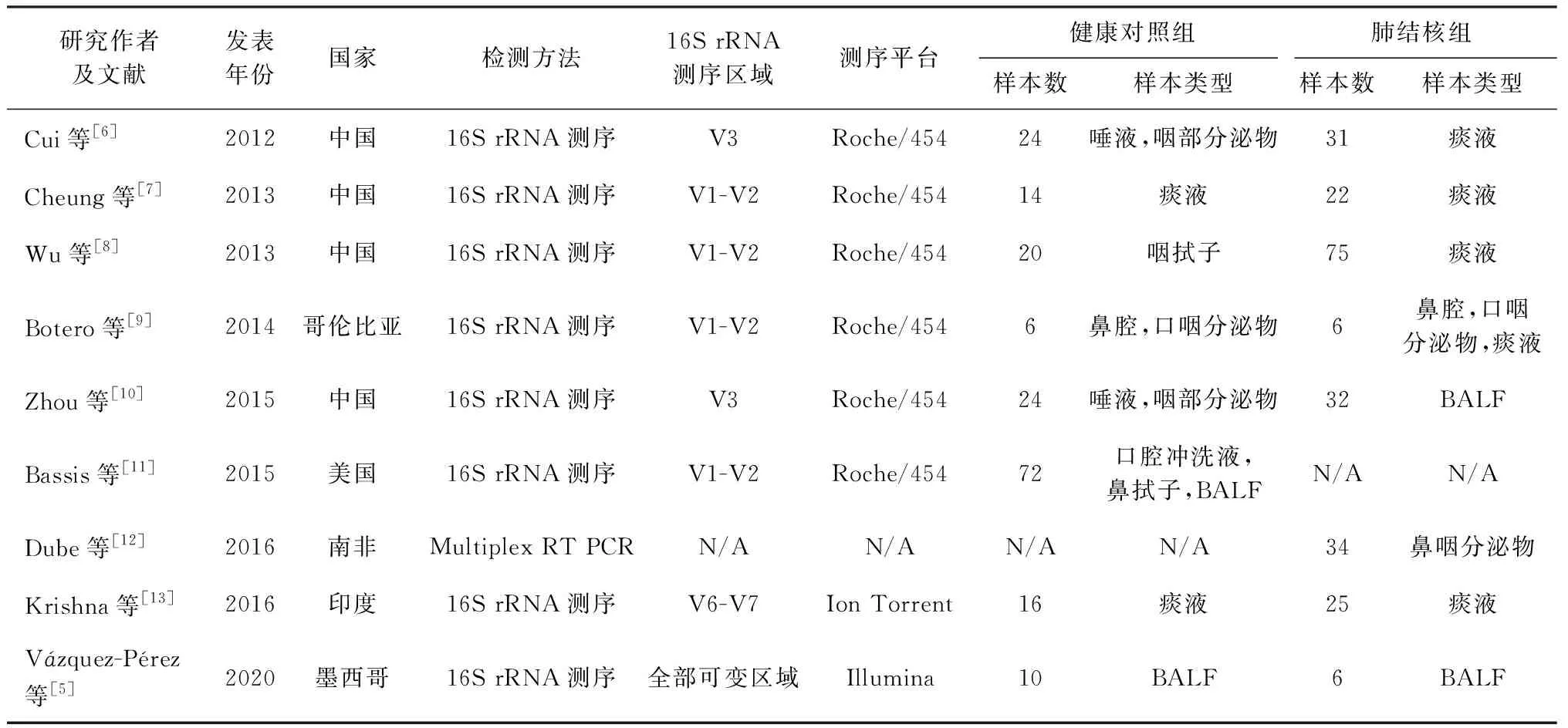
表1 纳入研究的基本信息

每个结点的注释用颜色范围表示;CTL:健康对照组;TB:肺结核组图2 iTOL可视化系统发育树

图3 肺结核组和健康对照组菌群门水平物种组成图
三、肺结核患者和健康人群呼吸道菌群α多样性分析
通过样本的α多样性分析可以反映微生物群落的物种丰富度和均匀性。α多样性分析的分析指数有观测到的OTU数目(估算物种相对丰度)、Chao1(估算物种丰富度)、Faith PD指数(基于物种间进化关系估算物种丰富度)、Evenness指数(估算物种均匀度)和Shannon(考虑物种丰富度和均匀度)指数。由图5可见,肺结核组观测到的OTU数目明显高于健康对照组(286.60±22.82和42.88±2.49),差异有统计学意义(P=0.00016)。图6结果显示,肺结核组Chao1指数高于健康对照组(266.50±92.71 和38.44±2.86),但差异无统计学意义(P>0.05)。图7显示Faith PD指数在两组之间差异明显(13.57±2.58和6.52±0.22;P=0.00056),说明肺结核组在亲缘差异上较健康对照组更高。虽然肺结核组的Evenness指数无明显变

图4 肺结核组和健康对照组呼吸道菌群属水平物种组成图
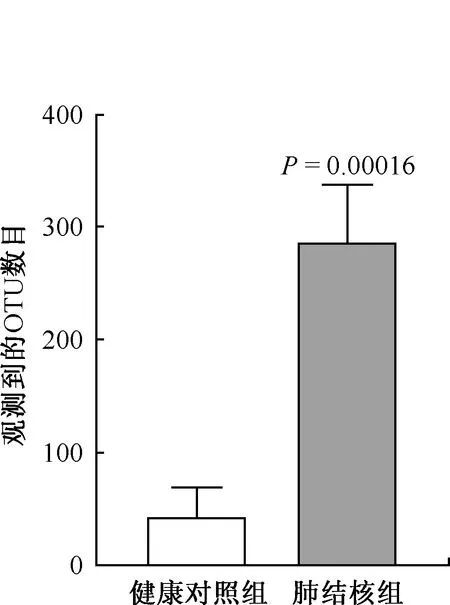
图5 两组呼吸道菌群观测到的OTU数目(observed OTUs)比较

图6 两组呼吸道菌群Chao1指数比较

图7 两组呼吸道菌群系统发育多样性Faith PD指数比较

图8 两组呼吸道菌群Evenness指数比较

图9 两组呼吸道菌群Shannon指数比较
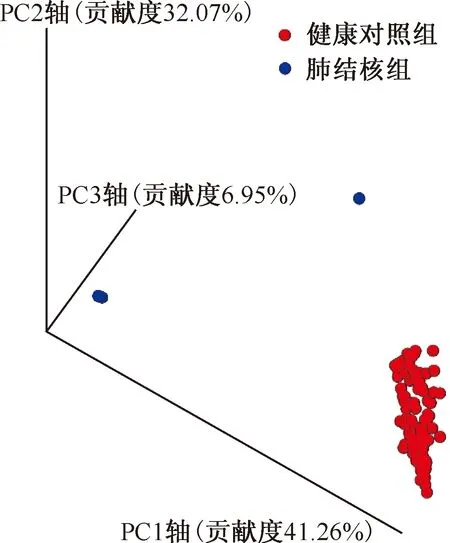
图10 两组呼吸道菌群PCoA分析(加权 Unifrac距离方法)

表2 两组呼吸道菌群ANCOM物种组成差异分析(门水平和属水平)
化(P>0.05,图8),但Shannon指数要明显高于健康对照组(7.17±0.24和4.42±0.07)(P=0.00016,图9)。综合5项指数分析可知, 肺结核患者呼吸道内菌群群落多样性要高于健康人群呼吸道内的正常菌群。
四、肺结核患者和健康人群呼吸道菌群β多样性分析
根据加权UniFrac距离和非加权UniFrac距离来进行的PCoA分析。分别用Jaccard距离、Bray-Curtis距离、未加权 UniFrac距离和加权UniFrac距离进行分阶段聚类算法达到ASV,提取每个ASV代表序列,得到样本的总体菌属在空间的分布图。由图10可知,PCoA分析显示在加权 UniFrac距离中,PC1轴贡献度为41.26%、PC2轴贡献度为32.07%,PC3轴贡献度为6.95%时可基本将肺结核组和健康对照组呼吸道菌群区分开,说明两组菌落组成有一定区别。对上述四种距离进行统计检验,在Jaccard、Bray-Curtis、未加权 UniFrac和加权 UniFrac 距离上,P值均为0.001,具有统计学意义,肺结核组和健康对照组呼吸道菌群β多样性在这4种距离上能显著区分开来。
五、群落差异丰度分析
肺结核组和健康对照组进行ANCOM群落丰度差异分析,在门水平,两组中丰度有差异的物种有6种,其中5种在健康对照组聚集,为Firmicutes、Actinobacteria、Bacteroidetes、Fusobacteria、Proteobacteria,另有OD1门在肺结核组聚集(表2)。属水平上,健康对照组有较大丰度差异的有Firmicutes-Bacilli-Lactobacillales-Streptococcaceae-Streptococcus,Bacteroidetes-Bacteroidia-Bacteroidales-Prevotellaceae-Prevotella,而肺结核组主要富集在OD1门下的属内(表2)。TM7门TM7-3纲CW040目F16科、Proteobacteria门Gammaproteobacteria纲、Fusobacteria门Fusobacteriia纲Fusobacteriales目、Firmicutes门Bacilli纲Lactobacillales目Leuconostocaceae科、Firmicutes门Bacilli纲Bacillales目Bacillaceae科是肺结核组特有的菌属。说明肺结核感染后,呼吸道内菌群发生了较大改变。
讨 论
存在于人体的微生物群包括细菌、真菌、病毒等,其中以细菌为主。自口腔、上及下呼吸道至肺,均有呼吸道菌群的存在,且菌群的数量及种类逐渐下降,变异也相应缩小[14]。因此,来自痰液和BALF标本能更好地代表远端气道。本文分析的测序数据来自5项研究,主要来源于痰标本和BALF标本,其余来自口咽、鼻咽的标本。基于临床上存在部分的无痰肺结核患者,而且健康人很难获取痰标本和BALF标本。因此,虽多种不同部位的样本增加了准确表述小呼吸道菌群的难度,但笔者综合5项研究而加大了样本量,或可减少部分偏倚,比单项研究的结果更加准确。
肺结核患者呼吸道菌群的特征和健康人不同[2]。笔者综合分析后发现肺结核患者呼吸道菌群的α多样性大于健康人群。8项有对照组的研究中,有3项研究和笔者的研究结果一致,3项研究提示肺结核组和健康组呼吸道菌群的α多样性相似,1项研究未涉及α多样性分析。不同的研究有不同的结果,考虑和各研究样本大小和测序深度的不同有关。另外,肺结核组与健康人群菌群结构也不一致。在β多样性评估中,未加权 UniFrac距离评估差异物种间的进化距离,不考虑物种丰度区别;加权 UniFrac距离同时考虑物种间的进化距离以及物种丰度的差异;Jaccard分析各物种的有无,而不考虑物种丰度,侧重于评估物种丰富度;Bray-curtis同时评估物种的有无以及相对丰度。本研究中,肺结核组与健康对照组能在组内聚类,且在Jaccard、Bray-Curtis、未加权 UniFrac和加权 UniFrac 距离上,P值均为0.001,差异具有统计学意义,提示两组呼吸道菌群在组成上有差异。此结果和纳入的8项病例对照研究结果均一致[5-10,12-13]。进一步分析该差异模型,有望开发出新的无创筛查方法,评估肺结核的发生、疾病控制水平。
菌群丰度分析结果提示,在门、属水平上,两组优势菌种种类不太一致,呼吸道菌群的改变不仅是菌群间相对比例失调,而且存在物种种类上的变化。本研究发现,在肺结核组和健康对照组中,Firmicutes、Proteobacteria、Bacteroidetes、 Actinobacteria、Fusobacteria等是主要的细菌门。然而,从统计学上看,两组上述菌门的相对丰度差异有统计学意义,且在健康对照组中更常见。此外,在肺结核组中发现了独特的细菌种类。F16科、Gammaproteobacteria纲、Fusobacteria门Fusobacteriales目、Firmicutes门下的Leuconostocaceae科和Bacillaceae科是肺结核特有的菌群种类。研究表明梭杆菌目在结核分枝杆菌感染6 d后被发现[15];在气溶胶感染结核分枝杆菌的动物模型中,梭杆菌目相对丰度上也发生了较大变化。这些研究结果为临床上结核病易并发真菌感染、革兰阴性菌感染提供了理论依据[16]。然而,目前对肺结核呼吸道菌群的研究极少,尚无菌群种类和肺结核及其并发肺部感染的发病机制的深入研究。深入探讨肺结核和健康人群特有的菌群种属,可能有益于结核病的病理基础、诊断和靶向治疗的新发展。
呼吸道菌群和结核感染可相互作用、互相影响[4]。正常情况下微生物菌群处于动态平衡状态,调节宿主的免疫反应,参与能量代谢,维护机体内环境稳定。目前在小鼠和人类研究中均明确了结核分枝杆菌感染可导致微生物菌群发生改变[15,17-18],我们的研究也证实了这一点:结核病患者的呼吸道菌群的数量、多样性及种类均发生了较大变化(图3~9)[14]。抗结核方案所用的抗生素或结核病的主要危险因素(HIV感染、营养不良、糖尿病、酗酒、吸烟和污染等)等均可致菌群失衡[19-20];此外,也有研究认为结核分枝杆菌感染可通过引起Th1和Th2免疫异常,间接造成菌群失衡[21]。反之,菌群失衡可使机体易感结核甚至发生疾病。现已发现菌群失衡导致结核病的致病机制可能有:(1)失衡的肺部菌群干扰免疫细胞亚群数量及功能,导致结核易感并可降低治疗反应[22];(2)通过肺-肠轴影响肠道菌群,进而影响营养及药物吸收和物质代谢,造成结核易感,影响治疗效果[22];(3)紊乱及失调的免疫细胞弱表达或不能表达重要病原体识别受体(巨噬细胞介导的C凝集素)和结核毒性细胞因子IFN-γ、TNF-α和IL-17,降低了对结核分枝杆菌的吞噬能力[23];(4)菌群失调消弱或耗尽了调节正常免疫功能的菌种,降低了机体对结核分枝杆菌感染的耐受能力[23];(5)使正常菌群代谢物短链脂肪酸、吲哚丙酸产生减少,影响免疫功能,造成结核分枝杆菌增殖。然而,目前对呼吸道微生物菌群与结核感染相互关系的了解仍处于早期阶段,微生物菌群的组成和功能如何影响结核分枝杆菌感染和结核病患病风险尚需要深入研究。
笔者对多项研究进行综合性描述及宏基因组学分析,初步表明与健康对照组相比,肺结核患者呼吸道菌群菌落特征和多样性发生了较大改变,并发现了肺结核患者特有的菌群种类。同时,本研究存在着一些不足,比如:纳入的研究论文中样本量较小,多为单中心研究,导致结果有发表偏倚的潜在风险(Egger检验的P=0.08)。未来需要进行大样本、多中心研究,以深入了解呼吸道菌群在结核病发生和局部免疫中的作用及影响。了解健康人群及肺结核患者呼吸道菌群的动态变化,可能为肺结核及其并发肺部感染的发病机制提供线索,并可能有助于设计出对结核病患者和公共卫生具有潜在直接有益影响的结核病治疗方案。
猜你喜欢
传染病信息(2022年3期)2022-07-15
保健医苑(2022年5期)2022-06-10
医学概论(2022年4期)2022-04-24
大众科学(2022年3期)2022-04-09
中华养生保健(2020年10期)2021-01-18
World Journal of Hepatology(2020年7期)2021-01-14
中华养生保健(2020年7期)2020-11-16
中华肩肘外科电子杂志(2019年4期)2019-08-24
海峡姐妹(2018年4期)2018-05-19
安徽医科大学学报(2015年9期)2015-12-16
