
利用CRISPR-Cas9技术和Cre/loxP系统构建猪伪狂犬病病毒gE基因缺失疫苗株
2020-09-27 04:06史志斌马宁宁刘占王永生陈陆
畜牧与兽医 2020年10期
史志斌,马宁宁,刘占,王永生,陈陆
(河南农业大学牧医工程学院动物传染病教研室,河南 郑州 450046)
伪狂犬病病毒(pseudorabies virus,PRV)属疱疹病毒科α疱疹病毒亚科[1],引起的猪伪狂犬病(porcine pseudorabies)严重危害全球养猪业[2]。猪急性感染PRV后引起妊娠母猪流产、产死胎及木乃伊胎、种猪不育、新生仔猪脑脊髓炎等[3-4]。急性感染耐过猪和隐性感染猪能建立终生潜伏感染,潜伏的病毒存在于三叉神经节、嗅球和扁桃体等组织,在应激状态下活化并向外排毒,因此,潜伏感染猪是伪狂犬病潜在传染源[5-7]。PRV潜伏感染特性给猪伪狂犬病的防控带来巨大困难。目前,防控本病主要依靠疫苗免疫,生产中常用的伪狂犬病疫苗主要是PRV毒力基因(如TK、gE、gI等)或糖蛋白基因(如gC、gG等)缺失的基因工程苗[8],尤其以TK和gE基因的缺失最为重要。gE基因缺失可降低PRV神经嗜性,也可作为诊断标志,通过对gE糖蛋白诱导抗体的血清学检测鉴别疫苗免疫猪和野毒感染带毒猪,为伪狂犬病的净化提供技术支持[9-11]。2012年始,伪狂犬病在我国传统疫苗免疫场再次出现暴发和流行,新出现的流行株表现毒力增强。为了研发针对当前流行株的疫苗,本文首先从实验室分离的4株PRV流行株筛选高免疫原性亲本株,然后利用CRISPR-Cas9技术和Cre/loxP系统对亲本株gE基因进行缺失,并对构建的gE基因缺失株制备的灭活疫苗的免疫效力进行评价,为伪狂犬病标志疫苗的研制提供参考。
1 材料与方法
1.1 主要试剂
EcoRⅠ、SpeⅠ、HindⅢ、BbsⅠ和T4DNA连接酶购自TaKaRa公司;低熔点琼脂糖购自北京索莱宝科技有限公司;2×DMEM高糖培养基购自杭州吉诺生物医药技术有限公司。
1.2 病毒和细胞
PRVHNQYY2012株、HNQXX2012株、HNQXY2012株、HNQBA2012株,Vero和293T细胞由本实验室保存。
1.3 质粒
pMD18-T载体购自TaKaRa公司;pX330和DH5α由本实验室保存;pcGlobin2-Cre质粒购自上海权阳生物科技有限公司;pEGFP质粒由本实验室合成保存。
1.4 实验动物
6周龄昆明小鼠购自河南省实验动物中心。
1.5 方法
1.5.1 引物设计
根据GenBank公布的PRV基因序列(登录号:KP722022.1),使用软件Primer 5.0设计gE基因上下游同源臂的扩增引物。利用http://crispr.mit.Edu设计靶向PRV gE基因的单链引导RNA(single guide RNA,sgRNA)。序列信息见表1。

表1 引物及sgRNA序列信息
1.5.2 PRV亲本株筛选
将HNQYY2012株、HNQXX2012株、HNQXY2012株、HNQBA2012株稀释至104TCID50/0.1 mL后灭活,病毒液和Montanide ISA 201 VG佐剂按体积比1∶1比例混合乳化,制成猪伪狂犬病灭活疫苗。25只昆明小鼠随机均分5组,1~4组分别皮下注射不同毒株制备的灭活疫苗,200 μL/只(104TCID50/只),最后1组注射DMEM培养基200 μL/只作对照。一免2周后进行二次免疫,一免2周、二免2周和二免4周时分别采血并测定中和抗体效价,选择抗体水平高、免疫原性好的毒株用于构建gE基因缺失株。
1.5.3 病毒基因组提取
HNQYY2012株接毒于Vero细胞,待细胞病变80%时收毒。酚氯仿法抽提病毒DNA后溶于TE,-20 ℃保存。
1.5.4 转移载体的构建
以HNQYY2012株基因组为模板,利用引物gEL-f/gEL-r和gER-f/gER-r分别扩增gE基因上、下游同源臂gEL和gER。PCR反应条件为95 ℃ 5 min;95 ℃ 30 s,60 ℃ 30 s,72 ℃ 80 s,30个循环;72 ℃ 10 min。gEL和gER分别用EcoRⅠ/SpeⅠ和SpeⅠ/HindⅢ双酶切,纯化后克隆到EcoRⅠ/HindⅢ双酶切的pMD18-T载体,获得重组质粒pTgE。pEGFP质粒经NheⅠ/SpeⅠ双酶切后,纯化获得EGFP片段并克隆到pTgE质粒上,得到重组载体pTgE-EGFP。
靶向PRV gE基因的sgRNA退火成双链,将pX330质粒用BbsⅠ酶切,形成黏性末端,经T4 DNA连接酶连接后,双链sgRNA与pX330质粒连接构成质粒pX330-sgRNA。
1.5.5 HNQYY2012ΔgE/EGFP+的获得和纯化
利用Lipofectamine®2000 Reagent转染。将HNQYY2012基因组、pX330-sgRNA和pTgE-EGFP按质量比1∶1∶2混于250 μL Opti-MEM,获得溶液A;250 μL Opti-MEM加入10 μL Lipofectamine®2000混匀,获得溶液B;溶液A和B轻缓混匀,室温静置20 min后缓慢滴加到293T细胞,36 h后-80 ℃冻融2次备用。处理后的细胞培养液(含细胞碎片)8 000 r/min离心5 min,收集上清液,经0.22 μm滤器过滤除菌并接种Vero细胞,PRV的基因组感染特性导致Vero细胞产生病变,达到70%时收获病毒,蚀斑纯化法挑取带绿色荧光空斑得到纯化病毒,命名为HNQYY2012ΔgE/EGFP+。
1.5.6 HNQYY2012ΔgE株的获得与纯化
共转染HNQYY2012ΔgE/EGFP+基因组和pcGlobin2-Cre质粒到293T细胞内,pcGlobin2-Cre质粒表达产生的Cre酶识别HNQYY2012ΔgE/EGFP+基因组EGFP基因两端同向的Loxp序列实现EGFP基因的敲除。参照1.5.5方法获得含HNQYY2012ΔgE基因组的上清液并接种Vero细胞。蚀斑纯化直至所有细胞病变均不带荧光时获得HNQYY2012ΔgE株。
1.5.7 HNQYY2012ΔgE株的PCR鉴定
酚氯仿法抽提HNQYY2012ΔgE病毒基因组,用引物gEL-f和gER-r进行PCR鉴定,并对PCR产物进行测序。
1.5.8 HNQYY2012株亲本株与ΔgE株一步生长曲线
HNQYY2012亲本株和HNQYY2012ΔgE以MOI=0.5接种Vero细胞,接毒后12、24、36、48、60、72 h收获病毒,测定TCID50并绘制一步生长曲线。
1.5.9 HNQYY2012ΔgE株LD50测定
35只昆明鼠随机均分成7组,1~3组分别皮下注射102、103和104的TCID50的PRV亲本毒,4~6组分别皮下注射102、103和104的TCID50的HNQYY2012ΔgE,第7组为DMEM的对照组。观察1周,Karber法计算LD50。
1.5.10 猪伪狂犬病灭活疫苗(HNQYY2012ΔgE株)的制备和免疫效力试验
HNQYY2012ΔgE株用Vero细胞增殖并定量为2×106TCID50/0.1 mL。0.3%的甲醛灭活后与Montanide ISA 201 VG佐剂按体积比1∶1乳化,制备成灭活疫苗。30只昆明鼠随机均分成6组,1~3组免疫灭活疫苗0.1 mL/只,4~6组为不做免疫的对照组。1~3组二免二周后和对照4~6组分别以5LD50、10LD50和20LD503种剂量攻毒,统计小鼠死亡情况。
1.6 统计与分析
利用SPSS 20.0对所得统计数据进行显著性分析,采用单因素方差分析进行显著性检验,多重比较采用新复极差法(SSR),设置显著性水平为0.05。最后用作图软件GraphPad Prism 6进行制图,数据表示的形式为“平均值±标准差”。
2 结果与分析
2.1 PRV亲本株筛选
如图1所示,在一免2周时4种毒株中和抗体水平均显著高于DMEM培养基对照组(P<0.05),但4种毒株之间差异不显著(P>0.05);在二免2周和二免4周时HNQYY2012和HNQXY2012株在抗体水平上相比其他毒株明显升高(P<0.05)。HNQYY2012株优于HNQXY2012株,因此选择HNQYY2012株为亲本株构建PRV gE基因缺失株。

同一处理内不同小写字母表示差异显著(P<0.05),相同字母表示差异不显著(P>0.05)
2.2 转移载体的构建
以pTgE为对照,用引物gEL-f/gER-r进行PCR扩增,pTgE-EGFP扩增产物长度大于对照组(图2),PCR产物测序结果表明扩增的条带包括gE上游同源臂、EGFP序列和gE下游同源臂,证明成功构建转移载体pTgE-EGFP。pX330-sgRNA进行双酶切鉴定,酶切后呈单一条带,未连接的PX330空载体酶切后出现2个条带(图3),证明成功构建了pX330-sgRNA质粒。

M.15 000 bp DNA Marker;1.pTgE;2.pTgE-EGFP

M.15 000 bp DNA Marker;1.pX330-sgRNA的EcoRⅠ和BbsⅠ双酶切;2.pX330的EcoRⅠ和BbsⅠ双酶切
2.3 重组病毒HNQYY2012ΔgE的获得与纯化
pX330-sgRNA作用下,HNQYY2012基因组和pTgE在293T细胞内发生重组,收获样品后接种Vero细胞,72 h后出现明显带绿色荧光的细胞病变,说明有重组病毒产生。经过7轮噬斑纯化,得到重组病毒HNQYY2012ΔgE/EGFP+(图4)。在Vero细胞上扩增HNQYY2012 ΔgE/EGFP+并提取基因组,以相同方法将HNQYY2012ΔgE/EGFP+基因组和pcGlobin2-Cre质粒共转染293T细胞,获得重组病毒HNQYY2012ΔgE基因组后再次接种Vero细胞,72 h后出现不带绿色荧光的细胞病变。相同方法纯化后获得纯度较高的重组病毒HNQYY2012 ΔgE。
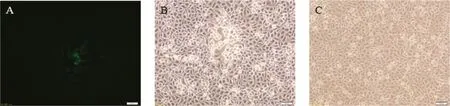
A.HNQYY2012ΔgE/EGFP+重组病毒接种Vero细胞;B.HNQYY2012ΔgE/EGFP+重组病毒接种Vero细胞;C.正常Vero细胞对照
以重组病毒HNQYY2012ΔgE和HNQYY2012ΔgE/EGFP+基因组为模板,用引物gEL-f/gER-r扩增gE基因。重组病毒HNQYY2012ΔgE/EGFP+扩增之后在4 400 bp的位置出现条带,而重组病毒HNQYY2012ΔgE在2 600 bp的位置出现条带(图5)。对重组病毒HNQYY2012ΔgE的PCR产物进行测序,结果表明HNQYY2012ΔgE重组病毒gE基因完全缺失。

M.250 bp DNA Marker;1.空白对照组;2.gEL-f/gER-r对HNQYY2012ΔgE的PCR扩增;3.gEL-f/gER-r对HNQYY2012ΔgE/EGFP+的PCR扩增
2.4 HNQYY2012ΔgE和亲本毒一步生长曲线的测定
由图6可知HNQYY2012ΔgE和亲本毒的最高滴度分别为106.8TCID50/0.1 mL和107.0TCID50/0.1 mL,差异不显著(P>0.05),HNQYY2012ΔgE的病毒滴度和亲本毒差异不大。在24、36、60 h时间点,亲本毒滴度极显著高于HNQYY2012ΔgE(P<0.01或P<0.001),说明gE基因缺失对PRV的滴度和增殖速度影响较小。

**P<0.01,***P<0.001
2.5 HNQYY2012ΔgE和亲本毒LD50测定
测定HNQYY2012ΔgE和亲本毒LD50分别为103.8TCID50和102.5TCID50,差异显著(P<0.05),说明gE基因缺失后PRV的毒力有一定程度下降。
2.6 灭活疫苗(HNQYY201ΔgE株)的免疫效力试验
灭活疫苗(HNQYY2012ΔgE株)免疫小鼠对5LD50、10LD50剂量攻毒保护率均为100%,20LD50的攻毒剂量组有1只小鼠存活,保护率为20%,3组对照组保护率均为0。结果表明猪伪狂犬病HNQYY2012 gE基因缺失株灭活疫苗具有良好免疫效力。小鼠生存曲线如图7所示。

图7 HNQYY2012ΔgE灭活疫苗免疫小鼠攻毒后生存曲线
3 讨论
CRISPR-Cas9基因编辑技术是从细菌和古细菌中发展演变而来,可以对靶基因进行敲除和突变,其技术成熟,成本较低,在活体动物、细胞和病毒中都有广泛的应用[12-14]。徐广军等[15]利用CRISPR-Cas9技术建立了GTP蛋白酶M(IRGNs)基因缺失的MDCK细胞系。IRGNs是由干扰素诱导细胞产生的,在抵御病原微生物中发挥着核心的作用。通过敲除MDCK细胞的IRGMs,可以进一步研究IRGMs基因在细胞内抵御病原微生物感染中的作用。吴涛等[16]利用CRISPR-Cas9基因编辑技术对肠道病毒71型(EV71)的VP4区域进行了剪切,从而抑制了EV71病毒的复制,为治疗EV71提供了新的途径。Whitworth等[17]利用CRISPR-Cas9技术分别敲除了猪的CD163和CD1D基因,这2个基因被认为是猪繁殖与呼吸综合征病毒(PRRSV)的受体基因和抗原递呈因子,通过敲除这2个基因后,猪体对PRRSV具有良好的抗病能力,证明CRISPR-Cas9可以成功地应用到动物疫病防控策略中。Cre-loxp系统也常被用于进行病毒基因组重组的系统。梁苑燕等[18]首先构建出了带有loxp位点和EGFP的PRV gE-/EGFP+株,之后通过Cre酶处理后,得到了不带EGFP的PRV gE-株,之后又利用同样的方法得到了PRV gE-/TK-株。Xu等[19]利用CRISPR-Cas9和Cre-loxp系统同时对PRV的gE/TK基因进行缺失,成功构建了PRV gE-/TK-株。本试验利用CRISPR-Cas9技术和Cre-loxp系统对PRV gE基因进行缺失,经过PCR和测序鉴定,证明gE基因得到完全缺失。试验结果表明,利用CRISPR-Cas9技术和Cre-loxp系统进行病毒的基因缺失是可行且高效的,同时以2012年后新出现的高免疫原性PRV流行株为亲本株构建的gE基因缺失株并制备灭活疫苗更符合市场需求。
HNQYY2012ΔgE株和亲本株的一步生长曲线结果比较发现,两者的增殖最高滴度差异不显著,但gE基因的缺失导致PRV在细胞上的增殖速度减缓。Wang等[11]研究发现HN1201株感染PK-15细胞后病毒增殖速度减缓却可以达到相近的病毒滴度,病毒蚀斑变小,与本试验结果一致。HNQYY2012ΔgE毒力和亲本毒相比差异显著,半数致死量高于亲本毒表明gE基因缺失后PRV毒力下降。gE基因是PRV的主要毒力基因,在病毒的复制及病毒在细胞间的传播过程中起到关键的作用。研究证明gE基因缺失后,PRV的毒力明显的降低[20]。将HNQYY2012ΔgE株制备灭活疫苗,并在昆明鼠上进行了免疫效力试验,结果表明,免疫灭活疫苗后,小鼠可以抵御105TCID50剂量的亲本毒的攻击,而对106TCID50剂量攻毒后的保护率明显降低。结果证明所制备的灭活疫苗可以对小鼠产生有效的保护。HNQYY2012ΔgE株灭活疫苗免疫动物,取得的血清中不含gE抗体,可用于鉴别疫苗免疫动物和野毒感染动物,故本试验获得的毒株可作为PRV的候选疫苗株使用。
猜你喜欢
小天使·一年级语数英综合(2022年2期)2022-03-30
长江蔬菜(2021年12期)2021-04-04
动漫星空(兴趣百科)(2020年3期)2020-03-24
中国果业信息(2019年11期)2019-01-05
湖南畜牧兽医(2016年3期)2016-06-05
西南农业学报(2016年5期)2016-05-17
兽医导刊(2016年12期)2016-05-17
兽医导刊(2016年12期)2016-05-17
浙江农业科学(2016年11期)2016-05-04
中国卫生产业(2015年10期)2015-03-11
