
2010-2019年某CRO公司药物临床试验项目盲态审核中方案偏离的相关数据分析
2020-09-21 08:46董玉君王蓉余秋钿程国华
中国药房 2020年17期
关键词:质量管理
董玉君 王蓉 余秋钿 程国华

摘 要 目的:研究方案偏离对药物临床试验结果的影响,为提高药物临床试验的质量管理水平提供参考。方法:收集广州某合同研究组织(CRO)公司2010-2019年药物临床试验的盲态数据审核表,分析方案偏离总体特征、“722公告”(指国家食品药品监督管理总局2015年7月22日发布《关于开展药物临床试验数据自查核查工作的公告》)前后的方案偏离情况以及方案偏离对全分析集(FAS)人群划分的影响,并提出相应建议。结果:最终纳入45个试验项目,涉及454个中心、14 304例病例和5 562例次方案偏离。最常见的方案偏离类型依次是超窗、违背纳入与排除标准、脱落,分别占方案偏离的36.88%、20.71%、18.43%。不同试验分期和不同药物类型的项目发生方案偏离的程度无统计学意义(P>0.05),而“722公告”前后不同阶段项目发生方案偏离的程度有显著性差异(P<0.05);“722公告”后的超窗、违背纳入与排除标准和服药依从性的偏离发生率有所升高。发生方案偏离的病例有82.07%可以进入FAS,纳入FAS且未进入符合方案集(PPS)的人群占总体偏离人群的53.99%,其中脫落、合并用药偏离分别占19.51%、4.29%,服药依从性偏离的人群均未进入PPS。结论:脱落、违背纳入与排除标准、超窗是造成临床试验方案偏离的主要因素。“722公告”对药物临床试验人员的质量管理意识的提高起到了一定的促进作用。建议应选择恰当的统计方法控制偏倚,从试验设计、人员培训及机构管理建设各方面加强药物临床试验质量管理,减少方案偏离的发生。
关键词 方案偏离;药物临床试验;盲态审核;质量管理
ABSTRACT OBJECTIVE: To study the effects of protocol deviation on the results of clinical trials, and to provide reference for rising the quality management of drug clinical trials. METHODS: Blind data review forms for clinical trials of a contract research organzation (CRO) company in Guangzhou from 2010 to 2019 were collected to analyze general characteristics of protocol deviation, the situation of protocol deviation before and after the “722 announcement” (Announcement on Carrying Out Self-inspection and Verification of Drug Clinical Trial Data issued by CFDA on July 22, 2015) as well as the effects of protocol deviation on full analysis set (FAS) population division. The suggestions were put forward. RESULTS: A total of 45 trials were included, involving 454 centers, 14 304 disease cases and 5 562 cases of protocol deviation. The most common types of protocol deviations were over-window, violation of criteria of the inclusion and exclusion, and drop-out, which accounted for 36.88%, 20.71% and 18.43% respectively. There was no statistical significance in protocol deviation degree of clinical trials with different stages or drug types (P>0.05); there was significant difference in the degree of protocol deviations in clinical trials with different stages before and after the “722 announcement” (P<0.05); the incidence of deviations from over-window, violation of cirteria of the inclusion and exclusion, and medication compliance had increased after the “722 announcement”; 82.07% of cases with protocol deviations could enter FAS, and the population who included in FAS but did not enter per protocol set (PPS) accounted for 53.99% of the total deviation, of which deviations from drop-out and combined medication accounted for 19.51% and 4.29% respectively. All cases with deviation from medication compliance did not enter PPS. CONCLUSIONS: Drop-out, violation of criteria of the inclusion and exclusion, and over-window are the main factors that cause clinical trial protocol deviations. The “722 announcement” played a certain role on improving the quality management awareness of the personnel in drug clinical trial. Appropriate statistical methods should be selected to control bias, and to strengthen the quality management of drug clinical trials and reduce protocol deviations, by paying attention to trial design, staff training, institutional management and construction.
KEYWORDS Protocol deviation; Drug clinical trial; Blind review; Quality management
任何有意或无意偏离或违反《药物临床试验质量管理规范》(GCP)或试验方案的行为被称作方案偏离(Protocol deviation)或方案违背(Protocol violation)[1]。当方案偏离积累到某种程度时将会影响试验的有效性和安全性评价结果。盲态审核是临床试验数据管理的关键步骤,是数据管理与统计分析衔接的桥梁[2]。试验结束到揭盲之前,主要研究者、医学总监、生物统计学家、数据管理员和申办者会在盲态的情况下对数据质量进行核对评估,对偏离方案的人群是否进入全分析集(Full analysis set,FAS)、符合方案集(Per protocol set,PPS)、安全性分析集(Safety set,SS)進行讨论,以便确定统计分析计划[3]。对盲态审核中方案偏离情况的分析和评价是研究结果科学可靠的必要保证。
为保证临床试验数据的真实性、可靠性和完整性,国家食品药品监督管理总局(CFDA)于2015年7月22日发布了《关于开展药物临床试验数据自查核查工作的公告》(以下简称“722公告”)[4],该公告规定已申报生产并且待审的新药申请以及相关药物临床试验项目均需申请人对临床试验数据进行自行检查。这是我国临床试验数据核查理念变革的关键节点,代表着我国药企、合同研究组织(Contract research organization,CRO)与药物临床试验机构将积极推动临床试验的合规性。为了解我国临床试验管理中常出现的问题,进一步完善我国药物临床试验法规与指导原则,本研究以某CRO公司的盲态数据审核表中临床试验项目的试验分期、药物种类、所处“722公告”前后时期作为分类标准,归纳项目所出现的偏离类型,分析偏离方案中的人群划分情况。通过偏离类型、方案偏离的差异性及对试验结果的影响分析,反映临床试验质量控制中存在的问题,提出改进措施,为药物临床试验的质量管理提供参考,促进临床试验高质量地完成。
1 资料与方法
1.1 研究对象
收集广州某CRO公司2010-2019年完成药物临床试验项目的盲态数据审核表。由于Ⅰ期临床试验主要为药动学(PK)或药效学(PD)分析,偏离情况与其他分期差异较大,不易于合并分析,故本研究不纳入Ⅰ期临床试验项目。
1.2 研究方法
1.2.1 收集试验项目基本信息 收集项目的临床试验分期、药物类型、试验时间、病例数、中心数等信息。本研究中的不同临床试验分期包括Ⅱ、Ⅲ、Ⅳ和未分期临床试验,不同药物类型包括中药、化学药及生物制品;另外以“722公告”为时间节点,将试验项目分为“722公告”前结束的项目(以下简称“722公告”前)、“722公告”前启动且“722公告”后完成的项目(以下简称跨越“722公告”)和“722公告”后启动的项目(以下简称“722公告”后)三个阶段。
1.2.2 分析项目方案偏离总体特征 按照盲态数据审核表的方案偏离情况进行划分,定义超窗、违背纳入与排除标准、脱落、合并用药、未完成相应检查、服药依从性等不同方案偏离的类型。将参加试验的受试者的偏离情况进行分析、归类、量化、统计,将结果记录于数据提取表中。方案偏离的类型定义见表1。
1.2.3 分析“722公告”前后的方案偏离情况 对“722公告”前后的方案偏离情况进行亚组分析,比较“722公告”前、跨越“722公告”和“722公告”后三个阶段的项目所发生的方案偏离情况。
1.2.4 分析方案偏离对FAS人群划分的影响 FAS是最大程度将入组的受试者都纳入统计分析的数据集,对于已经被分配随机号的受试者以最合理的方式最少地剔除,一般只有在发生重大方案违背、误纳、未服用过试验药物或随机分配后无任何观测数据的情况下才不纳入FAS[5]。PPS是FAS中更加符合方案的病例。分析盲态数据审核表中方案偏离对FAS的影响以及方案偏离对纳入FAS的人群是否纳入PPS的影响,再结合方案偏离情况进一步分析对试验疗效结果的影响。
1.3 统计学方法
采用SPSS 20.0软件对所得数据进行分析。计数资料以率表示;通过集中趋势和离散趋势进行正态分布检验,非正态分布的计量资料以中位数与四分位间距M(P25,P75)表示。不同试验分期和试验时间所发生方案偏离的差异用多个样本比较的Kruskal-Wallis检验分析,不同药物类型发生方案偏离的差异用两样本比较的Mann-Whitney检验分析。P<0.05表示差异有统计学意义。
2 结果
2.1 纳入项目的基本情况
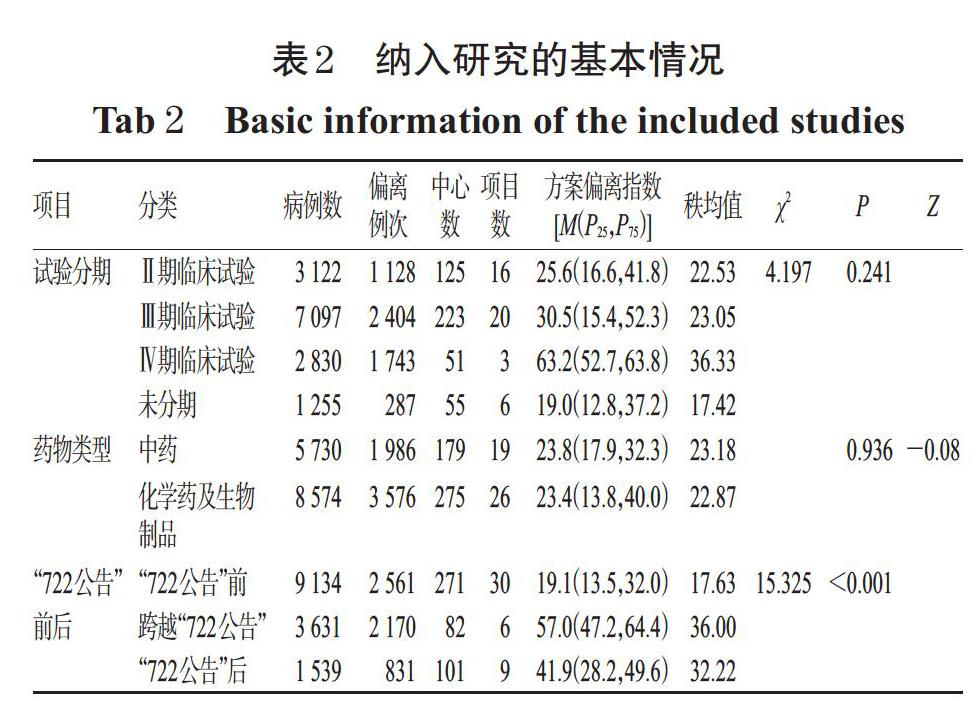
通过归类分析,最终纳入45个符合条件的临床试验项目,共涉及454个中心、14 304例病例及5 562例次方案偏离。纳入研究的基本情况见表2[表中方案偏离指数代表每100个病例发生偏离的例次数,方案偏离指数=100×(偏离病例次数/病例数)。
由表2可知,分别对不同临床试验分期、不同药物类型、“722公告”前后的偏离方案的差异性进行分析,结果显示,不同试验分期和不同药物类型的临床试验项目发生方案偏离的程度没有显著性差异(P>0.05);“722公告”前、跨越“722公告”和“722公告”后临床试验项目的方案偏离指数的差异有统计学意义(χ2=15.325,P<0.001),可认为三个阶段方案偏离的程度显著不同。
2.2 对盲态审核中临床试验方案偏离类型的总体分析
方案偏离常见类型的分布情况见表3。
由表3可知,临床试验中最常见的方案偏离类型依次是超窗、违背纳入与排除标准、脱落,分别占总体偏离情况的36.88%、20.71%、18.43%。
2.3 “722公告”前后偏离方案的亚组分析
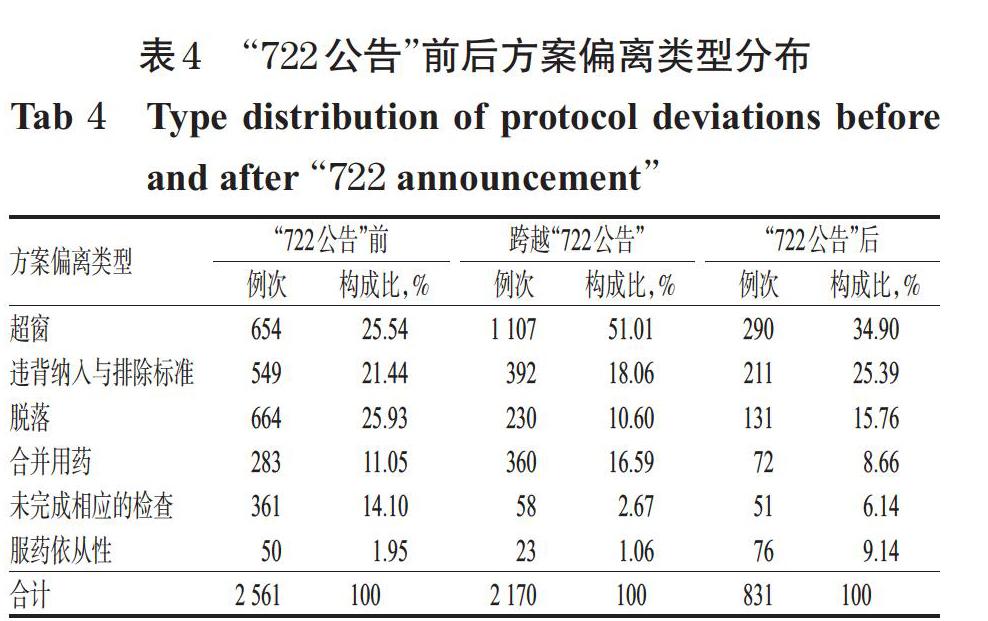
以所处“722公告”前后时期为分类标准,对比“722公告”前、跨越“722公告”以及“722公告”后三个阶段方案偏离的分布情况,结果见表4。
由表4可知,“722公告”前后药物临床试验方案偏离情况的分布存在差异:跨越“722公告”期间的临床试验项目出现超窗偏离的比例高达51.01%,“722公告”后期开展的临床试验项目出现超窗、违背纳入与排除标准和服药依从性等偏离的比例均大于“722公告”前结束的试验项目所出现的情况。
2.4 方案偏离对FAS人群划分的影响
本研究入选的项目中可纳入FAS的方案偏离有2 507例次,FAS中未进入PPS的方案偏离例次占53.99%,其中脱落、合并用药和服药依从性偏离例次分别占19.51%、4.29%和2.00%。而进入与未进入PPS的超窗和违背纳入与排除标准的方案偏離例次比例相当。方案偏离中纳入FAS中的PPS情况如表5所示。
3 讨论与建议
3.1 方案偏离类型的分析
超窗是临床试验中最常出现的问题,访视时间的延后或提前会引起高估或低估药物的疗效。根据超窗对试验结果的影响程度可分为严重超窗与轻度超窗两种类型:严重超窗是指受试者关键检查或访视日期远超出规定的时间窗口,会明确影响试验药物的有效性评价,如果涉及到主要疗效指标严重超窗的情况,最后会从FAS集中剔除;轻度超窗是时间窗在可容忍的范围内,依从性符合要求或非关键检查超窗,不影响有效性评价。超窗通常由受试者或者研究者依从性不佳引起[6],如受试者对试验不配合、态度不重视或研究者对试验方案设计不恰当、不熟悉。本研究出现的超窗多由研究者间接造成,因此试验前应对研究者进行全方位的培训,增强研究者的GCP意识和对方案的熟悉程度,研究人员在访视前也需提前通知受试者。
违背纳入与排除标准也是临床试验中常出现的问题,多为研究人员对方案执行意识的薄弱导致误纳入,以及由于纳入受试者病情较为复杂,在试验期间发生不可控的生理或病理变化引起。遵守纳入与排除标准、筛选合格的受试者是药物临床试验顺利开展的前提条件。筛选受试者时应去除病情过于复杂、可能影响疗效评价、不适合参与试验的人群,一般包括妊娠妇女、肝肾功能不全、对试验药物可能过敏或合并其他重大疾病等人群[7-8]。方案执行前,研究者需对方案的科学性以及可操作性进行评估。在对临床监查员(CRA)和临床协调员(CRC)进行培训时,应重点强调纳入与排除标准,指出易违背的关键点,要求其严格按照方案执行,研究者及CRC需对相关结果认真核对,减少病例误纳入的可能性。在试验启动会上,还应向研究者强调试验操作应以方案要求为准,对试验过程中的疑问需及时记录并反馈给CRA或者项目组成员。
脱落为临床试验中较易发生的问题之一,可能会引起临床试验的疗效指标和安全指标缺失,产生结果偏倚。病例脱落与受试者的依从性存在较大的相关性。受试者常因外出打工、交通不方便、换工作、搬家、家人反对或怀疑试验药物的效果而要求退出或者失访。受试者的年龄、性别、受教育程度、性格、成长环境、病情程度不同,其对临床试验的依从性也会有所差异。研究者在筛选受试者时应多方面评估受试者的依从性,优先选择依从性良好的受试者;试验方案的设计方面,尽可能减少访视点,简化试验流程,增加方案的可操作性[9]。CRA访视时要关注受试者的用药情况,通过查看病历报告、溯源医院信息系统、门诊开药信息,来确认合并治疗情况;一旦发现受试者合并使用方案禁止的其他药物,须及时记录,并报告方案偏离。
本研究表明,大部分偏离原因与质量保证和质量控制有关。在药物临床试验中,应加强第三方稽查、研究者监督、项目组成员质控及CRA的监查等,发生质量问题后及时采取纠正措施;对于多中心发现的问题应进行汇总分析,若项目存在共性问题,可对同类问题采取预防措施,降低同类型问题再次发生的概率。
3.2 方案偏离的差异性分析
“722公告”后,CFDA不断完善临床试验相关法律法规,颁布临床试验数据管理有关的指导规范与指南。在借鉴国际规范、技术指南并结合我国现状的基础上,CFDA于2016年发布《临床试验数据管理工作技术指南》《药物临床试验的生物统计学指导原则》《药物临床试验数据管理与统计分析的计划和报告指导原则》《临床试验的电子数据采集技术指导原则》,以确保药物临床试验数据的规范性、完整性和真实性。2017年6月1日,CFDA成为国际人用药品注册技术协调会(International Council for Harmonization,ICH)成员,加入ICH是国际对我国药品监管水平和能力认可的标志,也意味着对我国临床试验的质量提出了更高的要求[10]。
本研究结果显示,“722公告”后,超窗、违背纳入与排除标准及服药依从性偏离情况的发生率升高,但这一趋势不代表“722公告”后开展的临床试验项目实际发生的偏离率较之前高。“722公告”核查公告的原则为自查纠错从宽、被查处理从严、严惩故意造假、允许规范补正,专家组成员需在此原则下开展现场检查工作,确保临床试验数据的真实、完整与可靠性,确保受试者的安全和权益受到保护。面对有史以来最严的数据核查要求,大部分项目被撤回或整改,以往被忽略的违背方案或者漏报的试验项目纷纷上报。跨越“722公告”阶段的项目正好面临整改阶段,方案偏离被发现并且上报的数量要高于“722公告”前开展的项目。可认为“722公告”政策出台后,研究者主动报告、自查等因素致使被发现并且上报的方案偏离更接近实际情况,发布药物临床试验数据核查公告后相关人员的GCP意识有所提升,对质量的控制更加规范,更加关注数据的真实性和完整性。
3.3 方案偏离对临床试验结果的影响
临床试验包括优效性、等效性和非劣效性临床试验,FAS和PPS常用于试验药物的疗效性分析,两者在不同的试验中所起的作用不同[11]。由于FAS的分析结果比较保守,依从性差的受试者的加入可能导致药物疗效被低估,在优效性试验中宜采用FAS作为主要分析集。PPS可显示试验药物按规定使用的效果,相对于上市后的药物疗效,可能高估了实际疗效。在对等效性或非劣效性临床试验的再统计分析时,用FAS所得到的结果不一定保守,此时可以结合PPS的分析结果。
[ 4 ] 国家食品药品监督管理总局.关于开展药物临床试验数据自查核查工作的公告[EB/OL]. (2015-07-22) [2020- 03-02]. http://www.nmpa.gov.cn/WS04/CL2182/299803.html.
[ 5 ] 国家食品药品监督管理总局.药物临床试验的生物统计学指导原则[S]. 2016-06-01.
[ 6 ] 王晓霞,李育民,高瑾,等.药物临床试验中受试者的依从性问题研究[J].中国药物与临床,2009,9(6):507-508.
[ 7 ] 张强,蒙萍,单爱莲.关于药物临床试验方案中纳入、排除标准的若干思考[J].中国临床药理学杂志,2017,33(2):99-101.
[ 8 ] 李雪迎.临床实验设计三要素之研究对象[J].中国介入心脏病学杂志,2014,22(5):317.
[ 9 ] 陈国霞.浅谈新药临床试验中受试者依从性的管理[J]. 中国民族医药杂志,2016,22(3):55-56.
[10] 郭薇,谢林利,曹丽亚,等.加入ICH对我国药物临床试验机构工作的影响和思考[J].中国药房,2019,30(11):1445-1448.
[11] 李雪迎.临床试验研究统计学设计方法简述-优效性设计、等效性设计以及非劣效性设计[J].中国介入心脏病学杂志,2014,22(8):482.
[12] LESAFFRE E,DE K. Estimating the power of comp- liance-improving methods[J]. Control Clin Trials,2000,21(6):540-551.
[13] URQUHART DJ. Role of patient compliance in clinical pharmacokinetics[J]. Clin Pharmacokinet,1994,27(3):202-215.
[14] 张抗,李文元,冯硕,等.临床试验中脱落、退出和失访病例的统计学处理和报告规范[J].中医杂志,2016,57(14):1204-1207.
[15] CRUTZEN R,VIECHTBAUER W,KOTZ D,et al. No differential attrition was found in randomized controlled trials published in general medical journals:a meta-analysis[J]. J Clin Epidemiol,2013,66(9):948-954.
[16] VAN DLM,LORENZ TJ. Addressing missing data in cli- nical trials[J]. Ann Intern Med,2011,154(2):113-117.
[17] 衡明莉,陈丽嫦,王骏.临床试验中缺失数据处理方法研究[J].中国临床药理学杂志,2019,35(22):2948-2952.
[18] 张念先.临床试验常用缺失数据处理方法的局限性分析[J].中国新药与临床杂志,2009,28(9):659-662.
(收稿日期:2020-04-08 修回日期:2020-07-03)
(編辑:刘明伟)
猜你喜欢
人间(2016年27期)2016-11-11
现代企业文化·理论版(2016年14期)2016-10-21
中国市场(2016年36期)2016-10-19
科学与财富(2016年28期)2016-10-14
科学与财富(2016年28期)2016-10-14
科技视界(2016年20期)2016-09-29
科技视界(2016年20期)2016-09-29
