
B-cell lymphoma-2 inhibition and resistance in acute myeloid leukemia
2020-09-14 08:41
Lindsay Wilde,Sabarina Ramanathan,Margaret Kasner,Department of Hematology and Medical Oncology,Sidney Kimmel Cancer Center at Thomas Jefferson University Hospital,Philadelphia,PA 19107,United States
Abstract
Key words:Acute myeloid leukemia;B-cell lymphoma-2;Venetoclax;Metabolism;Leukemic stem cell;Resistance
INTRODUCTION
Acute myeloid leukemia (AML) is a heterogeneous group of aggressive hematologic malignancies characterized by the uncontrolled proliferation of genetically altered immature myeloid cells.Accumulated clonal leukemic stem cells (LSC) are inherently nonfunctional and arrested in differentiation causing rapid bone marrow failure and,if untreated,eventual death[1].
An estimated 21500 patients are diagnosed with AML yearly in the United States[2].Despite advances in molecular prognostication and therapeutic targeting,AML remains a significant cause of morbidity and mortality.The current 5-year survival rate remains <30%[3].Patients above age 65 and those with poor performance status,pre-existing comorbidities,or biologically aggressive disease have especially poor prognoses,as do patients who relapse after hematopoietic stem cell transplant[4].
Since the first publication by Yateset al[5]in 1973,the standard therapeutic approach for treating AML has relied upon intensive induction chemotherapy with the 7 + 3 protocol,a cytarabine and anthracycline based regimen.Individuals who were unable to tolerate intensive chemotherapy had few options[5].It has only been in the last decade that a myriad of new drugs have changed this paradigm and gained approval for the treatment of AML.
Despite improvements in the success of up-front AML therapy,treatment for relapsed disease remains a significant challenge.Relapse occurs due to the emergence of chemotherapy resistant leukemic stem cells[6].Over the past decade,much has been learned about the complexity of the metabolic and molecular transformations that LSCs undergo.Interestingly,some of the same metabolic dysregulations are seen in other malignancies including colon,breast,and prostate cancer[7].Whole-genome mapping and targeted sequencing of serial samples of leukemia cells from individual patients has led to the discovery of distinct metabolic aberrations that play a role in relapse and,in some cases,are targets for drug development[8].
Novel therapies such as venetoclax,a specific B-cell lymphoma-2 (Bcl-2) inhibitor,have triggered a paradigm shift in the approach to AML and reinvigorated discussions about the link between metabolism and cancer.Though the majority of patients respond to venetoclax-based treatment,the depth and duration of response remain inadequate[9].Thus,understanding the metabolic rewiring that allows treatment resistance to develop is crucial.This review summarizes Bcl-2 inhibition in AML with a focus on mechanisms of resistance to venetoclax,in particular those related to leukemic cell metabolism.
LEUKEMIC STEM CELL METABOLISM
During evolution from normal hematopoietic progenitors to LSCs,cells undergo significant alterations in metabolic pathways including glycolysis,amino acid metabolism,and fatty acid metabolism.Similar to normal progenitors,primitive LSCs retain the ability to self-renew and remain in the G0 phase,allowing them to escape eradication by cytotoxic chemotherapy,which targets actively dividing blasts[10].
Glucose metabolism
Leukemogenic cells exist in a stressful hypoxic microenvironment and,in response,upregulate certain energy producing conduits to meet proliferative demand.Enhanced glycolysis plays a prime role in LSC proliferation.Increased glucose flux is directed by activated oncogenes,particularly expression of BCR-ABL and MLL-AF9,along with overexpression of hypoxia inducible factor 1[11].These genes upregulate glucose transporter 1 receptor expression,thereby promoting glucose entry and subsequent phosphorylation by hexokinase.Increased levels of hypoxia inducible factor 1,hexokinase,and genes upregulate glucose transporter 1 are described in patients with relapsed AML with poor response to chemotherapy[12].In vivostudies of aggressive leukemia cells have demonstrated a correlation between high glycolysis flux and decreased levels of autophagy,an evolutionary intracellular degradation process that is bypassed by LSCs[13].
Historically,it has been thought that malignant cells preferentially use cytoplasmic anaerobic glycolysis as a major carbon source (the so-called Warburg effect) over mitochondrial oxidative phosphorylation (OX-PHOS)[14].However,metabolomic studies have shown that mitochondrial OX-PHOS may be upregulated in LSCs as an adaptive mechanism[15].Excess oxidative stress has been described in various hematologic malignancies as a critical factor in initiation and progression of disease.There is growing evidence showing that AML LSCs generate increased levels of reactive oxygen species (ROS) primarily driven by mitochondrial NADPH oxidase and other pro-oxidant mechanisms.Sallmyret al[16]suggest that acquired genetic changes in myeloid malignancies lead to DNA damage and defective repair by directly increasing ROS production.Certain genetic abnormalities in AML such as RAS,IDH1/IDH2 and fms-like tyrosine kinase 3 (FLT3)/ITD mutations can directly disturb ROS metabolism causing an eventual shift to amplified ROS production[16].
Interestingly,the majority of LSCs preferentially maintain a low ROS state due to their quiescent nature.These low ROS LSCs were isolatedex vivoand subject to gene expression studies using RNA sequencing methods.Remarkably,they displayed a uniform overexpression of the Bcl-2 protein without upregulation of other antiapoptotic members[17,18].
Glutamine metabolism
The non-essential acid glutamine can be metabolized by glutaminases to glutamate and then α-ketoglutarate,which can go on to fuel the tricarboxylic acid cycle in the mitochondria[19].To sustain high proliferative advantage,LSCs may adapt a metabolic preference for glutamine to drive biomass.This so-called glutamine addiction has been demonstrated in multiple studies and represents a potential target for anti-leukemic therapy[20-23].
A number of oncogenes and pathways work to potentiate glutamine addiction in AML,including FLT3.In fact,metabolomic studies reveal that FLT3 inhibited LSCs are impaired in their glycolytic function and fittingly switch to utilize glutamine as primary fuel.Therefore,this metabolic dependency on glutamine metabolism poses a potential therapeutic vulnerability when targeted with FLT3 inhibition[24].Concurrent reduction of glutamine and Bcl-2 inhibition are being studied to compromise mitochondrial energy production and induce apoptosis,respectively[23].
The mammalian target of rapamycin 1 (mTORC1) signaling pathway is involved in numerous cellular processes including metabolism,cell growth,and apoptosis.Moreover,it has been shown to play an integral role in LSC development and proliferation[25-27].Glutamine availability is a rate-limiting step for mTORC1;therefore,removal of glutamine accordingly inhibits mTORC1 signaling and may be another metabolic mechanism for the treatment of AML[28].
B-CELL LYMPHOMA-2 MEDIATED MITOCHONDRIAL APOPTOSIS
Control of cellular proliferation and apoptosis is deregulated in cancer cells.Mitochondria play an intrinsic role in programmed cell death through release of soluble proteins from the intermembrane space,a process called mitochondrial outer membrane permeabilization (MOMP).A group of over 20 specialized proteins,known as the Bcl-2 family,are the prime mediators of this process[29](Figure 1).
Apoptosis is tightly regulated by an intricate balance between pro-apoptotic Baxlike proteins (e.g.,BAX,BAK and BAD) and anti-apoptotic Bcl-2 like proteins (e.g.,Bcl-2,Bcl-XL,Bcl-W and MCL-1) which are predominantly localized in the mitochondria.Bcl-2 prevents apoptosis by inactivating BAX and BAK.Bcl-XL blocks apoptosis by rendering mitochondrial pores impermeable thus inhibiting cytochrome C release.BAX and BAK proteins promote apoptosis by simply opposing Bcl-2 and forming oligomeric pores essential in MOMP[30].
Each of these apoptotic proteins are structurally distinguished by four groups of Bcl-2 homology (BH) domain (1-4).Functionally these BH domains,specifically the“BH3-only proteins” (e.g.,BID,BIM,BAD,PUMA,NOXA and BIK/NBK),sense cellular stress,activate pro-death signals,and coordinate the activity of other Bcl-2 proteins[31].The binding of apoptotic proteins is highly selective:BAD binds exclusively to Bcl-2,Bcl-XL,and Bcl-W,NOXA to MCL-1 and A1,and BIM can bind to all anti-apoptotic members[32].Upstream of the intrinsic Bcl-2 pathway,PUMA serves as a critical mediator of cell deathviap53-dependent and independent activation of BAX,BAK and dismissing inhibition of Bcl-2 family proteins.Most BH3-only proteins exist in an ambiguous conformation and at relatively low levels.Chemotherapeutic agents induce activation of BH3 only proteins to overcome the anti-apoptotic threshold resulting in cell death[33].

Figure1 Diagram of the intrinsic apoptotic pathway.
In response to cellular derangement,BH3-only proteins concurrently inhibit antiapoptotic members and activate pro-apoptotic members,BAX and BAK.Intracytoplasmic signaling leads to transformation of BAX into homo-oligomers and translocation of the proteins into the mitochondrial membrane forming pores to induce MOMP.As a result,voltage dependent anion channels are unlocked facilitating release of cytochrome C into cytosol,binding to apoptotic protease-activating factor 1,apoptosome formation,caspase activation,DNA fragmentation,and ultimately cell death[30].
Mitochondrial response to pro-apoptotic members,a process known as “priming”,has been studied as a measure of sensitivity to chemotherapy.Artificial priming of myeloblast mitochondria with BH3-only proteins (BIM or BAD BH3-peptide)supported the hypothesis that Bcl-2 inhibition may be a powerful strategy in targeting AML cells.Analysis of poorly primed,chemo-refractory AML cells showed increased sensitivity to BAD BH3-peptide mediated killing with potential for BH3 mimetic benefit even in low-primed AML[34].Knowing the level and specificity of priming prior to treatment may help in predicting the synergistic action of chemotherapeutic agents and Bcl-2 inhibitors.This functional approach to predicting mitochondrial response to BH3 peptides,termed BH3 profiling,could distinguish alterations between AML myeloblasts and HSCs.Certain BH3 peptides used for profiling inhibit selective Bcl-2 family proteins (e.g.,BAD BH3 peptide indicates dependence on Bcl-2,Bcl-XL,or Bclw)[35].MOMP induced by targeting such peptides hints at specific dependence on certain anti-apoptotic proteins through which they inhibit cell death[36].
Human LSCs were first discovered to modify expression of death receptors (e.g.,FAS and TRAIL receptors) to evade apoptosis.LSCs with very immature phenotype of CD34+/CD38- were able to confer both chemotherapy resistance and decreased capacity to induce Fas-induced apoptosis[37].Prominently,alteration of the Bcl-2 mediated pro-survival pathway and variant expression of effector proteins (BAX and BAK) are potent methods employed by LSCs to inactivate death signals.Bcl-2 is normally expressed in early myeloid progenitors but downregulated during myeloid differentiation.However,transgenic studies have shown that overexpression of Bcl-2 protects LSCs from various apoptosis-inducing stimuli[38].Bcl-2 overexpression leads to increased LSC numbers in the bone marrow and enhanced colony formationin vitroandin vivo[39].Remarkably,the bone marrow stromal microenvironment may facilitate this mechanism.Leukemic blasts thrive by exhibiting a higher degree of Bcl-2 when co-cultured with stromal cells.It is therefore possible that eliminating Bcl-2 protein function can eradicate early LSCs[40].
TARGETING B-CELL LYMPHOMA-2 IN ACUTE MYELOID LEUKEMIA
In 2005,ABT-737,a high-affinity small molecule Bcl-2/Bcl-XL/Bcl-W inhibitor,demonstrated single agent mechanistic killing of lymphoma and various solid tumor cell lines.Later studies demonstrated effective killing of primitive CD34+/CD38-populations with independent and synergistic action of conventional chemotherapeutics.Remarkably,this disruption was specific to LSCs without apparent damage to normal HSCs[41].Certain LSCs with increased MCL-1 and phosphorylated Bcl-2 were unaltered by ABT-737,proposing a potential co-target to bypass resistance in AML[42].
Progenitor blasts and chemo-resistant LSCs are heterogenous and possess a certain degree of metabolic plasticity.As discussed,LSCs adapt to rely on OX-PHOS as their predominant source of carbon as suggested by high mitochondrial mass and increased oxygen consumption[43].Chemically blocking Bcl-2 causes prompt and severe impairment of OX-PHOS with the potential to cut off a major power source for LSCs[17].
Bcl-2 dependence has been described as a hallmark of multiple hematologic malignancies including AML.This led to the study of venetoclax (ABT-199),an oral Bcl-2 inhibitor,as a single agent and in combination with hypomethylating agents for the treatment of AML.Venetoclax is highly specific for Bcl-2 but also inhibits several other members of the Bcl family,including Bcl-W[17,44].Strong preclinical data was evidenced by a median IC50 of approximately 10 nmol/L,and mitochondrial apoptosis occurring within 2 h of exposure[45].
Venetoclax monotherapy was first studied in high-risk relapsed/refractory AML patients and was found to have an underwhelming overall response rate of 19%[46].Given these results,the success of the combination of venetoclax with a hypomethylating agent (HMA) (either 7 d of azacitidine or 5 d of decitabine) or lowdose cytarabine in newly diagnosed,elderly AML patients was somewhat unexpected.Studies have demonstrated 50%-70% response rates for combination therapy in this high-risk population[47,48].In addition,in the HMA + venetoclax study,median overall survival was increased by 17.5 mo (double that of an HMA alone)[47].These pivotal results led,in November 2018,to the FDA approval of the combination of venetoclax plus an HMA or low dose cytarabine combo for adults >75 years who are not candidates for intensive induction chemotherapy[44].Patients with mutations in FLT3,IDH1/2,or mutations in the nucleophosmin gene were noted to have the most favorable responses[47].
Interim results from a Phase II study of ten-day decitabine plus venetoclax were recently presented and build upon the results of these initial studies.In this heterogeneous cohort of patients,those with newly diagnosed de novo AML had a CR/CRi rate of 95%.Furthermore,80% of these patients became MRD negative and 90% were alive at 6 mo[49].
Unfortunately,retrospective results for venetoclax combination therapy in the relapsed/refractory setting have not been as promising,however,prospective studies are ongoing.Overall response rates in these patients,some of whom have been heavily pre-treated,range from 21%-64%[50,51].Patients with secondary AML and those whose AML harbors a TP53 mutation have the poorest responses[52].Identifying the reasons for the disparity between response rates in newly diagnosed and relapsed disease has been the focus of much investigation,and has centered on a discussion of leukemic cell metabolism.
MECHANISMS OF VENETOCLAX RESISTANCE
There is increasing interest in understanding the mechanisms underlying venetoclax resistance.Genomic and protein analyses of expanding clones of LSCs after venetoclax treatment have identified a variety of potential adaptive mechanisms,including alterations in leukemic cell metabolism.
Initial hypotheses about resistance mechanisms focused on alterations in BH3-family protein expression.Reductions in Bcl-2 expression have been shown to promote primary and acquired resistance to venetoclax by alternate pathway activation and upregulated expression of other anti-apoptotic proteins such as MCL-1 and Bcl-xL[53].A study of AML cell linesin vitroshowed a definite and inverse correlation with the ratios of Bcl-2/MCL-1 transcripts and venetoclax sensitivity suggesting the importance of MCL-1 effect on sensitivity[54].Similarly,Niuet al[55]demonstrated that Bcl-2/MCL-1 transcript ratio may represent a potential biomarker in predicting response[55].As such,methodical targeting of MCL-1 during venetoclax therapy may delay the acquisition of venetoclax resistance[56].However,MCL-1 upregulation is only part of the venetoclax resistance story.
To try and better understand the basis of resistance,Chenet al[57]performed a genome-wide CRISPR/Cas9 loss of function screen in venetoclax-sensitive and venetoclax resistant clones (VRCs).The analysis demonstrated that specific genes involved in mitochondrial physiology,namely CLPB with HAX1,contribute to development of VRCs.CLPB,also known as chaperonine,is a protein-coding gene thought to maintain mitochondrial integrity by preventing the release of cytochrome C following death stimulus.Loss of CLPB impairs mitochondrial structure thereby triggering defective OXPHOS and glycolysis.CLPB was notably upregulated in VRCs suggesting a potential dependency and an amenable target.Correspondingly,analysis of CLPB-deficient AML cells showed that they were more sensitive to venetoclax treatment[57].
Similarly,Sharonet al[58],performed a genome-wide CRISPR knockout screen to look for potential genes that could be inactivated to reestablish venetoclax sensitivity[58].Interestingly,a glycine-to-valine mutation at amino acid position 101 was not identified in the Bcl-2 gene of VRCs.This mutation was previously proposed as an acquired venetoclax resistance mechanism in chronic lymphocytic leukemia[59].Instead,multiple genes-DAP3,MRPL54,MRPL17,and RBFA- encoding key parts of the mitochondrial translation apparatus were identified.LSCs exposed to the bacterial mitochondrial ribosome inhibitors tedizolid and doxycycline,both alone and in combination with venetoclax,showed a depleted CD34+ fraction with combination therapy,but not with venetoclax alone,suggesting that pharmacologic inhibition of mitochondrial translation may overcome resistance[58].
Findings of a study by Pollyeaet al[60]demonstrated that deeper and more durable responses to treatment with venetoclax and azacitadine were due to effective eradication of OXPHOS dependence.Directin vitromeasurement of ETC complex II activity and SDHA glutathionylation in primary AML cells upon venetoclax and azacitidine exposure confirmed decreased glutathione levels and correlating reduction in ETC activity[60].However,Joneset al[61]showed that OXPHOS levels in the LSCs of patients with relapsed AML are not reduced after HMA and venetoclax exposure suggesting that altered metabolism is an escape route for LSCs[61].Further evaluation of these LSCs identified an increased reliance on fatty acid metabolism,which may be targetable.
OVERCOMING RESISTANCE WITH VENETOCLAX COMBOS
Clinically,combining venetoclax with one or more other agents may be the key to overcoming resistance;many studies of this kind are underway[62].A comprehensive list of is found in Table 1.
VENETOCLAX + METABOLIC INHIBITION
Exploiting dependency on OXPHOS concurrently with Bcl-2 inhibition is a potential therapeutic strategy.Preclinical combination of OPB-111077,an OXPHOS inhibitor,with decitabine synergistically hindered the proliferation LSCs with a tolerable side effect profile.Triplet therapy with OPB-111077 + HMA and venetoclax in AML cells increased apoptosis rates to a greater degree than exposure to single agent OPB-111077 or venetoclax[63].A Phase I study of the triplet is ongoing.
The OXPHOS inhibitor IACS-010759 is another small molecule with promisingin vivoandin vitroactivity in LSCs in AML cell lines.This agent binds and inhibits complex I of the electron transport chain (NADH ubiquinone oxidoreductase) and is being studied in a phase I study of patients with relapsed/refractory AML.Safety is yet to be established with dose escalation,but mechanistically this is a sensible combination strategy with venetoclax[64].
Metformin,a biguanide used in diabetes management,has shown potential for antileukemic activity by directly targeting electron transport chain complex I activity and inhibition of constitutive mTOR activation.This in turn induces AMPK-independent apoptosis.Promising combination strategies with chemotherapy or other targeted therapies have been described with all-trans retinoic acid,ABT-737 (Bcl-2 inhibitor)and sorafenib in acute promyelocytic leukemia,T-cell acute lymphoblastic leukemia,and FLT3-ITD positive AML[65].Given its mechanism of action,the combination ofmetformin with venetoclax may be effective.
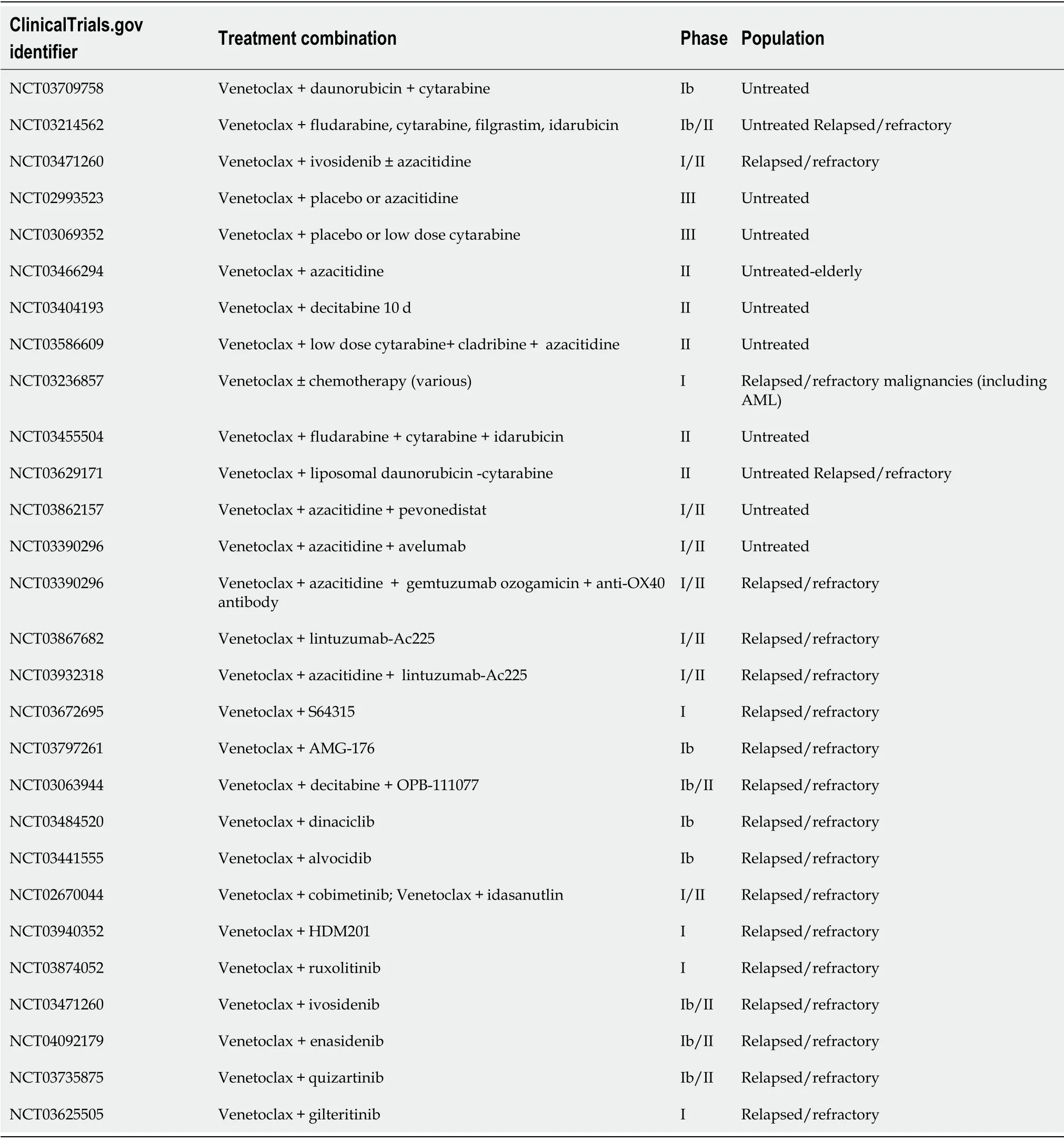
Table1 Clinical trials investigating venetoclax combination therapy
Finally,as discussed earlier,CLPB targeting can compromise mitochondrial matrix adding to Bcl-2 inhibition.Interestingly,a bacterial CLPB inhibitor has been developed and proposed as an antimicrobial agent with possible use in this setting[57].
VENETOCLAX+ DAUNORUBICIN/CYTARABINE
In vitrostudies conducted in AML cell lines and patient-derived AML samples have shown that venetoclax in combination with daunorubicin or cytarabine reduced MCL-1 protein levels resulting in increased DNA damage[66].Preclinical synergy translated to the clinical setting in an open label,multicenter trial study with 82 patients in which CR rate was 54% with a median OS of 10.1 mo.Lower response rates were observed for patients with prior hypomethylating agents[67].Investigations for Venetoclax with daunorubicin/cytarabine (7 + 3) and consolidation therapy are currently underway.
VENETOCLAX + MCL1 INHIBITOR/ CYCLIN-DEPENDENT KINASE 9 INHIBITION
MCL-1 inhibitors are under development to target VRCs.Direct MCL-1 inhibition with S63845 and A-1210477 plus venetoclax leads to synergistic cell killing of VRCsin vivoandin vitro.Additionally,several studies demonstrate preclinical synergy of A-1210477 and venetoclax where successful neutralization of MCL-1-dependent AML cells have been demonstrated[68].Dual inhibition of Bcl-2 and MCL-1 (with S55746 and S63845,respectively) has also shown strong activity against LSCs with relative sparing of normal progenitors.Researchers observed prolonged survival of xenograft models of AML with this combination[69].
More recent studies suggest synergy between venetoclax and inhibitors of Cyclindependent kinase 9 (CDK9),a transcriptional regulator of MCL-1,viaindirect targeting of MCL-1.Drivers of LSC survival like MCL-1 and MYC have very short half-lives making them expeditious targets to CDK9 inhibition.Alvocidib,aka flavopiridol,was the first of the CDK9 agents tested in combination with conventional chemotherapy[70].A newer agent voruciclib that inhibits CDK9,4,and 6 kinase diminishes transcription of MCL-1 downstream with better toxicity profile in comparison[71].
VENETOCLAX + MITOGEN ACTIVATED PROTEIN KINASE INHIBITION
Based on preclinical data,mitogen activated protein kinase pathway inhibitors such as cobimetinib (also a MEK1/2 inhibitor) have been studied with concomitant targeting of Bcl-2 in relapsed or refractory AML.Paduaet al[72]demonstrated disruption of the RAS/Bcl-2 complex in AML patient derived samples suggesting potential efficacy of the combination[72].Likewise,Hanet al[73]studied co-targeting of Bcl-2 and mitogen activated protein kinase in Bcl-2 protein enriched leukemic cells and synergistic killing was appreciated with over 60% growth inhibition in AML samples,including VRCs[73].Preliminary phase 1B clinical trial results,however,revealed increased gastrointestinal toxicity,mainly diarrhea,associated with cobimetinib[74].Newer MAP kinase inhibitors with better safety profiles are currently under development.
VENETOCLAX + PHOSPHATIDYLINOSITOL-3 KINASE/ MAMMALIAN TARGET OF RAPAMYCIN 1 INHIBITION
Dual Bcl-2 and phosphatidylinositol-3-kinase (PI3K/AKT) inhibition may help overcome both acquired and intrinsic venetoclax resistance requiring and is being evaluated in AML[75].Co-administration of venetoclax and apitolisib (GDC-0980:PI3K/mTOR inhibitor) or taselisib (GDC-0032:p110β-sparing PI3K inhibitor)induced profound cytochrome C release and apoptosis in various AML cell lines.AKT/mTOR inactivation and MCL-1 downregulation were also noted,with BAX and BAK mediated apoptosis of a CD34+/38-/123+ population while sparing the normal HSCs.
VENETOCLAX + MOUSE DOUBLE MINUTE 2 ANTAGONIST
Small molecule mouse double minute 2 homolog (MDM2) antagonists reactivate the tumor suppressor function of wildtype-p53 leading to downstream stimulation of proapoptotic BAX and NOXA.Further apoptotic pathways are promoted,like PUMA and BAD,to stabilize and degrade MCL-1.Studies with a combination of Nutlin-3a,a firstgeneration MDM2 inhibitor,and ABT-737,a Bcl-2 inhibitor,published a decade ago displayed durable induction of mitochondrial apoptosis of AML cells by the combination[76].Given preclinical rationale,researchers tested the combination of Bcl-2 and MDM2 inhibition (by idasanutlin) in wildtype-AML to boost activity of venetoclax and prevent upfront resistance[77].Safety and efficacy of venetoclax and idasanutlin has been studied in 39 patients with relapsed refractory elderly AML patients.Overall response rate was 46% with superior responses in IDH1/2,RUNX1,JAK2,MPL,and CALR mutations.TP53 and FLT3 mutations were associated with primary or secondary refractoriness[78].Additionally,updated data in both safety and efficacy appears to show reasonable tolerance to MDM2 and Bcl-2 inhibition.
VENETOCLAX + JAK2 INHIBITION
JAK inhibitors may combine with venetoclax to counteract bone marrow stromamediated resistance in AML.Cytokines activated by JAK/STAT signaling like GMCSF support AML cell proliferation and switch dependency of Bcl-2 to Bcl-XL[79].Correspondingly,ex vivostudies of isolated AML blasts expressed sensitivity to venetoclax + ruxolinitib combination as an effective method of killing[80].
VENETOCLAX + IDH INHIBITION
The small molecule IDH inhibitors enasidenib (IDH2) and ivosidenib (IDH1) are FDA approved for the treatment of AML.Inhibition of altered IDH1 and IDH2 enzymes along with hypomethylated genes can allow differentiation of LSCs[81].Studies investigating safety and tolerability of IDH1 and Bcl-2 inhibition are currently ongoing with ivosidenib and venetoclax,respectively[82].
VENETOCLAX + FLT3 INHIBITION
Sequencing studies were performed to assess the combination of venetoclax and the small molecule FLT3 inhibitor quizartinib in specific FLT3 ITD mutated xenograft models.The combination induced durable tumor regression for up to 3 mo after cessation of treatment[83].However,Chylaet al[84]noted that FLT3-ITD or PTPN11 mutations may confer intrinsic and acquired resistance to venetoclax[84].Clinical trials evaluating venetoclax and FLT3 inhibitor combination therapy are ongoing[9].
CONCLUSION
Up-front AML treatment with venetoclax in combination with a hypomethylating agent has shown impressive responses in multiple trials.Unfortunately,response durations are variable and patients still inevitably relapse.Attempts at identifying the cellular and molecular changes that occur after exposure to venetoclax have provided insight into mechanisms of resistance,namely alterations in LSC metabolism.Improved techniques to understand mitochondrial adaptations and the stromal microenvironment may aid in designing new therapeutic strategies.With more potent BH3 mimetics in development and rational combination therapies under investigation,the right strategy for building on the success of venetoclax treatment in AML is within reach.
 World Journal of Clinical Oncology2020年8期
World Journal of Clinical Oncology2020年8期
- World Journal of Clinical Oncology的其它文章
- GOECP/SEOR clinical recommendations for lung cancer radiotherapy during the COVID-19 pandemic
- Combination drug regimens for metastatic clear cell renal cell carcinoma
- Circular RNA and its potential as prostate cancer biomarkers
- Statins in risk-reduction and treatment of cancer
- Novel molecular targets in hepatocellular carcinoma
- Effectiveness of a novel,fixed dose combination of netupitant and palonosetron in prevention of chemotherapy induced nausea and vomiting:A real-life study from India