
粳稻分蘖数全基因组关联分析及候选基因的挖掘
2020-09-11 05:39:14张继峰刘华东王敬国刘化龙孙健杨洛淼贾琰吴文申郑洪亮邹德堂
中国农业科学 2020年16期
张继峰,刘华东,王敬国,刘化龙,孙健,杨洛淼,贾琰,吴文申,郑洪亮,邹德堂
(东北农业大学寒地粮食作物种质创新与生理生态教育部重点实验室,哈尔滨 150030)
0 引言
【研究意义】水稻是世界上主要粮食作物之一,随着全球人口的增长和耕地面积的日益减少,水稻每年需要达到 1%的增产才能满足人类对食物的需求。在经历矮化育种和杂种优势2次突破性飞跃后,水稻增产达到了瓶颈。长期栽培实践证明,杂种优势与理想株型相互利用是实现水稻进一步增产的重要途径。分蘖数是水稻理想株型的重要组成部分之一,也是水稻产量形成的重要性状。分蘖数决定有效穗数,适量的分蘖数会有助于高产,分蘖数过多会导致营养物质的浪费,分蘖数过少会导致过少的有效穗数,因此,适量的分蘖数是决定水稻高产的重要条件。【前人研究进展】水稻分蘖由分蘖节上各叶腋下处的分生组织发育而来,水稻分蘖形成包括2个部分,分别是分蘖芽的形成和向外生长过程。植物遗传学家现在已经克隆了MOC1、MOC2、MOC3、LAX1、LAX2和DTE1等与分蘖芽形成相关的基因。MOC1编码一个GRAS家族的核蛋白,主要在腋芽和叶腋原基表达,在营养生长和生殖生长阶段控制叶腋分生组织形成,moc1突变体表现为少蘖,同时花序轴和小穗减少[1-2]。MOC2/FBP1编码果糖-1,6二磷酸酶,该基因缺失表现为单孽、矮化、叶色变淡、每穗实粒数减少等,moc2突变体与moc1突变体不同,可以生长分蘖芽,但是分蘖芽的生长受到阻碍,不能正常发育成分蘖,果糖-1,6二磷酸酶是一个胞质FBP酶,该酶失活会导致植株蔗糖供应不足,从而导致moc2缺失突变体分蘖芽受到抑制[3]。MOC3/OsWUS/TAB1编码一个WOX蛋白家族成员,是一个转录抑制因子,主要在地上部愈伤组织中表达[4],该基因正向调控腋芽发育,可能是通过促进水稻同源异型基因OSH1的表达,进而在茎端分生组织中发挥重要作用[5],moc3-1突变体和moc2突变体一样,在分蘖芽发育过程中遭受破坏[3-4],Tab1/wus突变体表现为不能形成腋芽,导致没有分蘖[5]。LAX1编码一个植物特有的bHLH转录因子,该转录子含有一个碱性螺旋-环-螺旋(bHLH)结构域,该基因在地上部分顶端分生组织与新形成的分生组织之间的边界区域表达,LAX1蛋白在叶腋分生组织中积累,被 mRMA和随后的蛋白转运进行严格调控,该步骤对叶腋分生组织建立发挥重要作用[6-7]。LAX2/Gnp4编码一个核蛋白,和LAX1蛋白互作共同参与调控水稻腋生分生组织形成,进而调控水稻分蘖形态[8],Lax2与lax1突变体在营养阶段叶腋分生组织数目减少且大多数侧生小穗腋分生组织减少[8]。DTE1编码一个硼酸通道蛋白,在根的外层皮以及成熟叶片节区域表达,在硼饥饿处理下,dtel突变体表现出分蘖数增加、生长缓慢和育性下降等生长缺陷[9]。研究表明,大部分基因均通过双亲分离群体(RIL、NIL和BIL等)进行遗传位点挖掘,但鉴定的分蘖数遗传位点受到双亲遗传差异或分子标记密度的限制,所能挖掘鉴定的位点相对有限。全基因组关联分析以连锁不平衡(linkage disequilibrium,LD)为基础,通过群体基因型数据与表型数据检测有效关联位点,可以实现对同一复杂性状多数基因座以及一个基因座上多个复等位基因的检测[10-11]。同时,全基因组关联分析基于自然群体,不需要构建专门的作图群体,并且自然群体经过天然杂交多代,重组更加充分,定位精度更高。在人类和动物研究的基因挖掘中,关联分析是重要的手段,并且已在植物基因定位中得到广泛应用。2005年,Science杂志首次报道了人类年龄相关的黄斑性病症全基因组关联分析的研究[12]。受到人类遗传疾病研究的启示,植物遗传学家也将关联分析应用于作物遗传育种中,并取得了重要研究进展,为挖掘水稻有效等位基因提供了新方法。随着基因分型与测序技术的日益进步,目前,对于水稻的粒型、株高、抽穗期和剑叶形态等很多农艺性状开展了GWAS分析,同时也对病虫害抗性等生物胁迫和盐碱耐受性等非生物胁迫进行了全基因组关联分析,检测出许多重要的显著性SNP位点及候选基因,解析了许多重要性状的遗传基础[13-22]。【本研究切入点】尽管已有很多关于水稻分蘖数QTL/基因的研究报道,但大多是通过突变体基因定位或反向遗传学的方法进行研究,所挖掘到的QTL/基因有限。【拟解决的关键问题】本研究以来自不同国家和地区的295份温带粳稻品种为试验材料,结合高通量重测序获得的788 396个高质量多态性SNP,对粳稻分蘖数进行全基因组关联分析,鉴定影响粳稻分蘖数稳定存在的主效QTL,并对QTL区间内所有基因进行单倍型分析,根据基因注释筛选候选基因,为分子辅助育种提高产量提供理论基础。
1 材料与方法
1.1 试验材料以及性状考察
试验材料由国内外广泛收集的295份粳稻品种构成,其中,国内材料主要来自黑龙江省、吉林省和辽宁省等,国外材料主要来自日本、韩国和俄罗斯等(电子附表1)。所有材料于2018—2019年种植在东北农业大学阿城水稻试验基地,每份材料种植8行,每行20株,行、株间距为30 cm×13.3 cm,单株插秧。田间管理依照常规大田生产的基本方法。为了减少边际效应对表型的影响,于水稻分蘖盛期,在每个品种的第4行,选取表型相似的连续5株,考察每株水稻分蘖数,并计算平均值。
1.2 全基因组重测序
使用植物基因组DNA提取试剂盒提取295份粳稻品种的DNA,将检测合格的样品DNA采用Illumina HiSeq XTen进行高通量测序。利用GATK软件工具包进行SNP检测,使用Plink软件从初步获得的群体SNP中筛选出最小等位基因频率(minimum allele frequency,MAF)大于 5%,缺失率(missing rate)小于20%的788 396个SNP用于后续分析[23]。
1.3 群体结构与亲缘关系分析
利用 ADMIXTURE软件计算每个品种的遗传成分,将K值设置为1—10,运行结束后,提取每个K值的交叉验证(cross validation,CV)值,最小CV值所对应的K值为最优亚群分组数,所用群体被划分为3个亚群[23]。另外使用GCTA软件[24]进行主成分分析(principal component analysis,PCA),利用R语言绘制三维 PCA图。利用 MEGA 7[25-26]的邻接法(neighbor-joining)构建系统进化树(NJ树)。使用TASSEl 5.0软件[27]对群体材料间的亲缘关系进行评估[23]。
1.4 粳稻分蘖数全基因组关联分析
利用TASSEl 5.0软件的混合线性模型(mixed linear model,MLM),结合群体SNP基因型数据,进行粳稻分蘖数的全基因组关联分析,对GEC软件计算出的有效独立SNP数目进行阈值的确定,最终将P<5.46×10-6作为阈值,判定SNP标记与目标性状关联的显著性。利用R语言的“CMplot”包绘制曼哈顿图和 Q-Q图。依据每条染色体 LD衰减结果[23],以GWAS检测到的峰值SNP所在染色体上下游LD衰减距离区域界定为该QTL的染色体区间。
1.5 候选基因单倍型分析
对2年环境下共同检测到的QTL内所有基因进行单倍型分析。具体操作过程如下:从3KRGP的Rice SNP-Seek Database网站[28]中提取位于QTL区间内所有基因外显子区非同义突变SNP。利用之前研究中筛选得到的788 396个高质量SNP在位于该QTL区间内的所有SNP与3KRGP中提取的所有非同义突变SNP进行比对,确定其是否为非同义突变。若基因存在非同义突变,则先利用DnaSP软件对该QTL区间内所有存在非同义突变的基因进行单倍型分析,对不同单倍型(≥15份材料)的表型值进行多重比较,筛选不同单倍型表型值之间存在显著性差异的基因。对于不同单倍型表型值之间差异不显著的基因,再用启动子区(ATG前1.5 kb)的SNP进行单倍型分析。结合基因注释预测可能的候选基因。
2 结果
2.1 295份粳稻材料分蘖数表现
2018和2019年295份粳稻品种的分蘖数均具有较大的表型分布,分别为7.00—49.00个和6.00—33.80个(表1和图1),且2年总趋势基本一致,分蘖数平均值分别为18.35和15.75个。
2.2 全基因组关联分析
利用TASSLE 5.0软件的MLM模型分析,2018年在水稻第 8、9和 10染色体共检测到 3个 QTL(qTiller8、qTillerq9和qTiller10),2019年在水稻第9染色体检测到1个QTL(qTiller9),其中,qTiller9在2年中均被检测到(表2)。依据第9染色体LD衰减距离(52 kb)[23],选取峰值 SNP所在染色体的上下游LD衰减距离区域,界定为该QTL的区间(表2),结果显示,qTiller9被定位于水稻第9染色体15.01—15.12 Mb,与已定位影响水稻分蘖数QTL(qNOT9-1)[29]位于相同区间。

图1 2年环境条件下分蘖数频次分布直方图Fig.1 Histogram of tillering number frequency distribution under 2-year environmental conditions

表1 2年环境下295份粳稻材料的分蘖数表型值Table 1 The phenotypic values of tillering number among 295 japonica rice varieties in two years
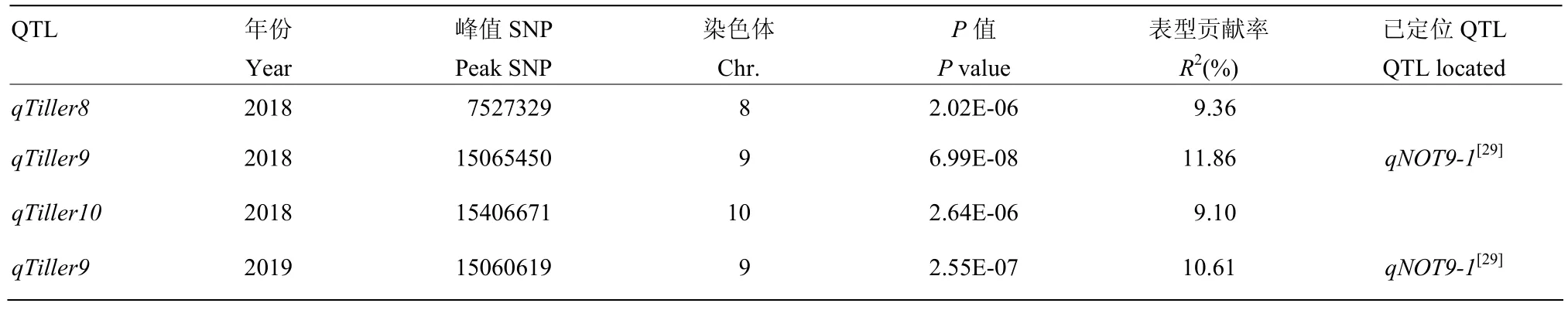
表2 粳稻分蘖数显著关联位点Table 2 Significant loci associated with tillering number in japonica rice
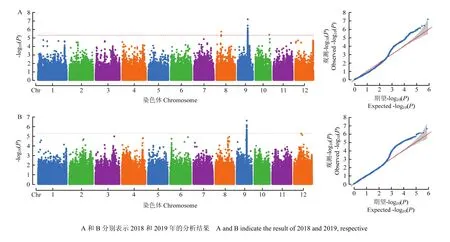
图2 分蘖数GWAS结果的曼哈顿图和QQ散点图Fig.2 Manhattan plots and quantile-quantile (Q-Q) plots of genome-wide association studies for the tillering number
2.3 候选基因分析
2018—2019年共检测到 3个影响粳稻分蘖数的QTL,其中,2年中能够稳定表达的QTL——qTiller9位于已定位影响水稻分蘖数QTL(qNOT9-1)区间内。因此,对qTiller9区间内全部15个基因的外显子区非同义突变SNP和启动子区SNP进行单倍型分析。结果显示,共有 6个基因(LOC_Os09g25090、LOC_Os09g25100、LOC_Os09g25150、LOC_Os09g25190、LOC_Os09g25200和LOC_Os09g25220)的不同单倍型分蘖数之间均检测到极显著差异水平(图 3)。LOC_Os09g25090被启动子区SNP分为2种单倍型,hap2(TAA)分蘖数极其显著大于 hap1(AGG);LOC_Os09g25100被非同义突变SNP分为2种单倍型,hap2(GAGA)分蘖数极其显著大于hap1(AGCC);LOC_Os09g25150被非同义突变SNP分为2种单倍型,hap2(ATG)分蘖数极其显著大于 hap1(GCC);LOC_Os09g25190被启动子区SNP分为2种单倍型,hap2(GCATCGCATCGACGCCGA)分蘖数极其显著大于 hap1(ATGCTGATGAAGTCATCC);LOC_Os09g25200被非同义突变SNP分为2种单倍型,hap2(TAG)分蘖数极其显著大于 hap1(AGA);LOC_Os09g25220被非同义突变SNP分为2种单倍型,hap1(GG)分蘖数极其显著大于 hap2(AA)(表 3和图3)。其中,LOC_Os09g25090和LOC_Os09g25100编码钙调蛋白依赖性蛋白激酶,它是脱落酸(ABA)表达所必需Ca2+的传感器。因此,推测LOC_Os09g25090和LOC_Os09g25100可能是影响水稻分蘖数的候选基因。

表3 候选基因单倍型分组及每种单倍型的SNP组成Table 3 Candidate gene haplotype group and the composition of each haplotype SNP

图3 候选基因不同单倍型之间分蘖数的箱线图Fig.3 Boxplots for the tillering number based on the haplotypes(hap) for candidate gene
3 讨论
水稻分蘖数是由多基因控制的数量性状,挖掘水稻分蘖数相关QTL和基因对水稻增产具有重要意义。连锁分析已经被证明可以有效地挖掘水稻分蘖数QTL,但是由于亲本数目较少,后代群体重组机会有限,因此,当等位基因在双亲中无差异时,QTL无法通过连锁分析检测出来。与连锁分析相比,全基因组关联分析以数百个品种组成的自然群体为研究材料,自然群体已经过天然杂交多代,重组更加充分,定位精度更高,并且能够同时检测同一基因座上的多个等位基因。本研究利用全基因组关联分析方法,于2018和2019年在水稻第8、9和10染色体共检测到3个 QTL,分别为qTiller8、qTiller9和qTiller10。HEMAMALINI等[29]利用IR64和Azucena杂交得到的56个随机DH系在水稻第9染色体11.80—15.55 Mb区间内定位到一个影响水稻分蘖数的 QTL(qNOT9-1),与本研究qTiller9位于相同区间,这验证了本研究结果可靠性的同时进一步缩小了 QTL区间,为后续候选基因分析提供了坚实的理论依据;qTiller8和qTiller10在前人研究中未见报道,且2个QTL仅在2018年检测到,需要通过遗传群体进一步验证。本研究进一步通过QTL所在的物理位置与前人已克隆基因进行比较,未发现QTL区间内有已知基因的报道。
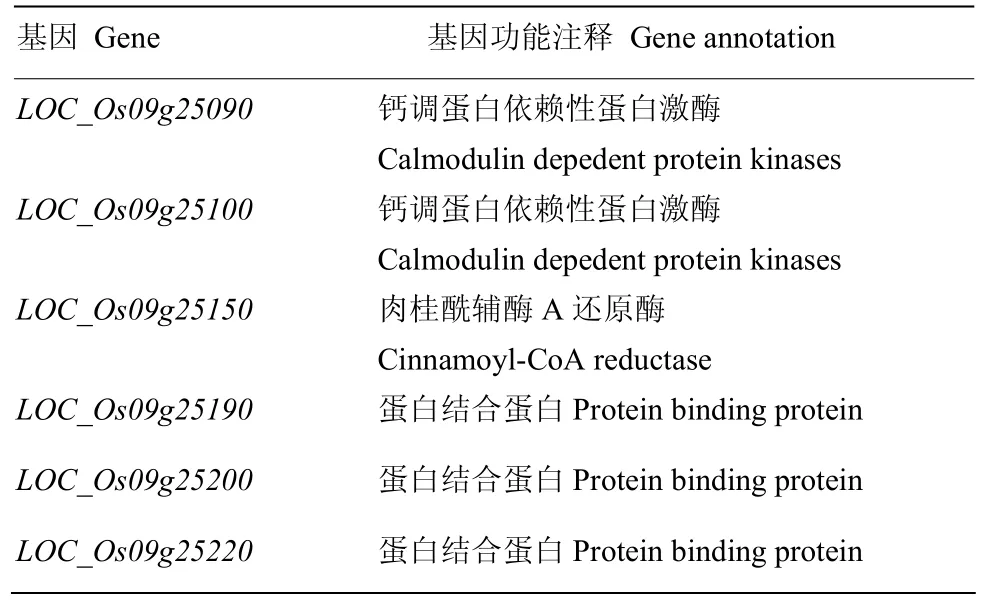
表4 候选基因的基因注释Table 4 Candidate gene of gene annotation
以往的全基因组关联分析挖掘相关性状候选基因,大多依据显著性峰值SNP位点上下游固定物理距离确定候选基因区域,然后通过基因功能注释进行候选基因的预测,然而水稻每条染色体的LD衰减距离各不相同,仅仅依靠固定距离确定候选基因区域,降低了该区域的精准性。本研究的检测到的QTL区域是依据每条染色体LD衰减距离及显著性峰值SNP位点确定的,这大大增加候选基因区域的精准性,同时本研究对共定位QTL区域内所有基因进行单倍型分析,再通过基因注释筛选单倍型表型值之间存在显著性差异的基因,这大大增加了候选基因筛选的效率及可靠性。
本研究通过对qTiller9区间内全部15个基因进行单倍型分析,发现有6个基因不同单倍型的分蘖数存在极显著差异。进一步通过基因功能注释筛选候选基因,发现6个候选基因分别属于钙调蛋白依赖性蛋白激酶、肉桂酰辅酶 A还原酶和蛋白结合蛋白。其中LOC_Os09g2509和LOC_Os09g25100均预测编码钙调蛋白依赖性蛋白激酶,其结构包括CaM结合域、丝氨酸/苏氨酸激酶域和3个EF手基序,是多个Ca2+传感器中的一员,可以检测到Ca2+的瞬时浓度[30-31],同时,Ca2+对脱落酸(ABA)的表达是必需的[37-39]。ZHU等[32]研究发现,9顺式-环氧类胡萝卜素双加氧酶OsNCED2是控制ABA合成代谢的关键酶,过表达OsNCED2的水稻转基因品系中,其分蘖数减少[33];另外,拟南芥基因MAX3和MAX4均编码类胡萝卜素裂解双加氧酶,是 ABA生物合成中的关键酶[34],拟南芥突变体max4存在一种侧芽抑制信号,该信号由根部向上运输,并与生长素互作进而抑制植株侧芽发生,这种信号的前体是类胡萝卜素,而植物中 ABA的前体同样为类胡萝卜素[35];STEVEN等[36]进一步研究发现,拟南芥中的max3和max4突变体是由MAX3和MAX4损伤引起的,导致2种突变体侧向分支急剧增加。以上研究表明,ABA在植物侧芽发生中发挥重要作用。本研究在水稻表达数据库(Rice Expression Profile Database)中查询LOC_Os09g25090和LOC_Os09g25100在田间整个生长生育时期各组织的时空表达量,发现2个候选基因在水稻茎部均有较高强度的表达。因此,推测2个候选基因可能在水稻分蘖处表达,通过调节Ca2+浓度介导ABA合成代谢进而影响水稻分蘖数。
4 结论
2018和2019年共检测到3个与粳稻分蘖数相关QTL(qTiller8、qTiller9和qTiller10),其中,qTiller9在 2年中均稳定表达,并与已定位影响水稻分蘖数QTL(qNOT9-1)重合。LOC_Os09g25090和LOC_Os09g25100可能为影响qTiller9的候选基因。
猜你喜欢
今日农业(2021年15期)2021-10-14 08:20:44
知识就是力量(2021年6期)2021-07-09 08:33:12
农民致富之友(2020年32期)2020-12-03 13:49:21
新课程·下旬(2018年7期)2018-01-19 11:55:40
河南农业(2017年7期)2017-07-25 09:22:05
生物学教学(2017年1期)2017-02-18 09:11:43
安徽医科大学学报(2016年12期)2017-01-15 14:21:44
山东农业工程学院学报(2016年6期)2016-12-01 05:38:19
浙江农业科学(2016年11期)2016-05-04 04:16:39
山东医药(2015年40期)2015-02-28 14:28:45
