
吡草醚悬浮剂和微乳剂的高效液相色谱分析方法
2020-09-11 08:38原万玲马杜康赵鹏跃黄啟良中国农业科学院植物保护研究所北京100193
现代农药 2020年4期
原万玲,马杜康,孟 璨,赵鹏跃,黄啟良(中国农业科学院植物保护研究所,北京 100193)
吡草醚(pyraflufen-ethyl),化学名2-氯-5-(4-氯-5-二氟甲氧基-1-甲基吡唑-3-基)-4-氟苯氧基乙酸乙酯,商品名速草灵、丹妙药、吡氟苯草酯等,由日本Nohyaku公司开发,是一种新型的苯基吡唑类苗后触杀型除草剂,其作用机理是抑制植物体内的原卟啉Ⅳ氧化酶,利用小麦与杂草对药剂吸收和代谢的差异,达到选择性地防治小麦田阔叶杂草的效果[1-2],也可有效促使成熟期的棉花脱叶[3]。2013年国内企业首次登记吡草醚产品。目前,我国登记的含有吡草醚有效成分的农药产品共9个,其中原药3个,母药1个,悬浮剂3个,微乳剂2个,应用于棉花和小麦田。然而,吡草醚农药产品尚无国家或行业标准,不能依法进行检验检测。因此,开展吡草醚农药产品中有效成分测定通用分析方法的研究,可以加快制定标准的步伐,尽早解决依法检验检测和依法监督管理问题。
有文献报道了以邻苯二甲酸二环己酯为内标物,利用气相色谱分析吡草醚原药的方法,该方法具有较好的线性、精密度和准确度[4]。刘同金等[5]采用气相色谱建立了吡草醚在小麦及土壤中的残留分析方法。秦旭等[6]采用高效液相色谱法测定了棉花中吡草醚的残留。张煜卓等[7]采用QuEChERS技术与气相色谱-串联质谱法测定了南国梨中吡草醚的残留。目前关于吡草醚制剂产品的分析方法尚未见报道。本文建立了在同一液相条件下测定吡草醚悬浮剂和微乳剂的方法,为吡草醚农药制剂产品质量检测提供依据。
1 材料与方法
1.1 仪器与试剂
Agilent 1260-DAD高效液相色谱仪(自动进样器),Chemstation工作站,美国Agilent公司;色谱柱:Agilent ZORBAX SB-C18不锈钢柱(250 mm×4.6 mm,5 μm);KQ3200B超声波清洗器,昆山市超声仪器有限公司;有机过滤器(微膜孔径0.45 μm)。
乙腈(色谱纯);二次蒸馏水;吡草醚标准品(质量分数99.97%),德国Dr.Ehrenstorfer公司;2%吡草醚悬浮剂,山东先达农化股份有限公司;2%吡草醚微乳剂,江苏龙灯化学有限公司。
1.2 液相色谱操作条件
流动相:乙腈+水(体积比65∶35);柱温:30℃;流速:1.0 mL/min;检测波长:243 nm;进样体积:5 μL;保留时间:吡草醚约9.5 min。
上述液相色谱操作条件是典型的操作参数,可以根据不同仪器特点和温度条件,对给定的操作参数进行适当的调整,以获得最佳效果。典型的吡草醚标准品、2%吡草醚悬浮剂、2%吡草醚微乳剂样品高效液相色谱图见图1、2、3。

图1 吡草醚标准品高效液相色谱图
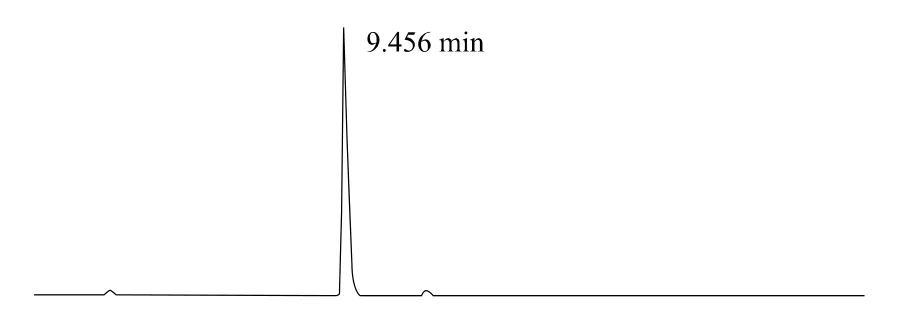
图2 2%吡草醚悬浮剂高效液相色谱图

图3 2%吡草醚微乳剂高效液相色谱图
1.3 测定步骤
1.3.1 标准样品的配制
称取0.025 g(精确至0.000 01 g)吡草醚标样于100 mL容量瓶中,加入20 mL甲醇,超声波振荡5 min,冷却至室温,用甲醇稀释至刻度,摇匀,记为标样溶液。
1.3.2 试样溶液的配制
分别称取2%吡草醚悬浮剂和2%吡草醚微乳剂试样0.3 g(精确至0.000 01 g)于100 mL容量瓶中,加入20 mL甲醇,超声波振荡5 min,冷却至室温,用甲醇稀释至刻度,摇匀,过滤待测。
1.3.3 测定
在上述操作条件下,待仪器稳定后,连续注入数针标样溶液,直至相邻两针吡草醚峰面积相对变化小于1.2%后,按照标样溶液、试样溶液、试样溶液、标样溶液的顺序进行测定。
1.3.4 计算
将测得的两针试样溶液以及试样前后两针标样溶液中吡草醚峰面积分别进行平均。试样中吡草醚质量分数w(%)按式(1)计算:

式中:w为试样中吡草醚质量分数,%;A2为试样溶液中吡草醚峰面积的平均值,mAU·s;m1为吡草醚标样的质量,g;P为标样中吡草醚质量分数,%;A1为标样溶液中吡草醚峰面积的平均值,mAU·s;m2为试样的质量,g。
2 结果与讨论
2.1 流动相的选择
由于2%吡草醚微乳剂中干扰较大,为使有效成分得到良好的分离效果,并且分析时间相对合适,峰形尖锐,保留时间适中,根据吡草醚的结构和性质,对不同配比的乙腈和水、甲醇和水流动相进行筛选。结果表明,流动相为乙腈+水(体积比65∶35),流速为1.0 mL/min,吡草醚保留时间为9.5 min,能够获得良好的分离效果。
2.2 检测波长的选择
利用紫外-可见检测器对吡草醚溶液在225~380 nm范围内进行扫描。吡草醚的紫外光谱图如图4所示。从图4中可见,吡草醚的最大吸收波长约为243 nm,在该波长处灵敏度较高,各种杂质不影响吡草醚的测定,能够满足分析的要求,故将检测波长确定为243 nm。
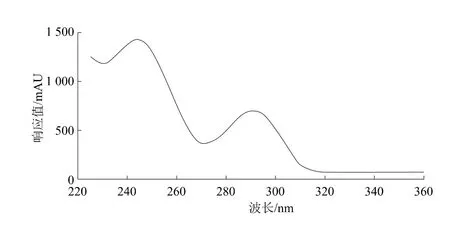
图4 吡草醚紫外吸收谱图
2.3 分析方法的特异性
本试验采用HPLC-DAD峰纯度分析法来鉴别吡草醚。吡草醚标样、2%吡草醚悬浮剂、2%吡草醚微乳剂中的吡草醚HPLC-DAD峰纯度均大于990,有效成分处无其他物质干扰,符合定量分析要求。
2.4 分析方法的线性关系
按1.3.1标准样品的配制方法配制标样溶液,用甲醇以1∶1的比例梯度稀释四次,获得5个浓度的有效成分线性相关溶液。待仪器稳定后,在上述操作条件下,按照分别进同样体积的标样,进行测定,取两次测定的平均结果。以吡草醚质量浓度为横坐标,峰面积为纵坐标绘制标准曲线。从图5可以看出,当吡草醚质量浓度在15.75 mg/L~251.92 mg/L之间(进样体积5 μL),与相应的吡草醚峰面积之间呈现良好的线性关系,计算得回归方程为y=8.732 8 x+9.456 2,相关系R2=0.999 9,完全可以满足定量分析要求。

图5 吡草醚标准曲线图
2.5 分析方法的精密度
从同一样品中称取5个试样,在上述色谱条件下进行分析,测得2%吡草醚悬浮剂和2%吡草醚微乳剂的标准偏差分别为0.07%和0.01%(表1)。

表1 分析方法的精密度试验结果
2%吡草醚微乳剂中吡草醚质量分数测定结果的变异系数为0.59,小于修改的Horwitz公式2(1-0.51ogC)×0.67=2.43(其中C为样品中有效成分质量分数的平均值),表明有效成分分析方法精密度的测定结果符合要求。
2%吡草醚悬浮剂中吡草醚质量分数测定结果的变异系数为0.73,小于修改的Horwitz公式2(1-0.51ogC)×0.67=2.42,表明有效成分分析方法精密度的测定结果符合要求。
2.6 分析方法的准确度
分别称取5个已知含量的悬浮剂和微乳剂试样于100 mL容量瓶中,加入1.3.1中配制的吡草醚标样溶液4 mL,用甲醇超声溶解,冷却至室温,用甲醇定容至刻度,摇匀。吡草醚的回收率按式(2)计算。
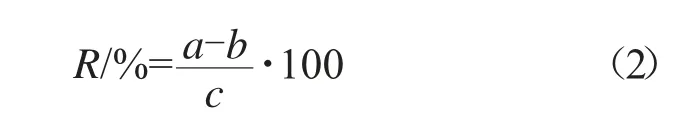
式中:R为回收率,%;a为测定量,mg;b为所称原药样品中待测组分量,mg;c为理论加入量,mg。
经测定,2%吡草醚悬浮剂和2%吡草醚微乳剂的回收率分别为97.45%和97.68%,试验结果见表2。表2表明有效成分分析方法准确度的测定结果符合要求。
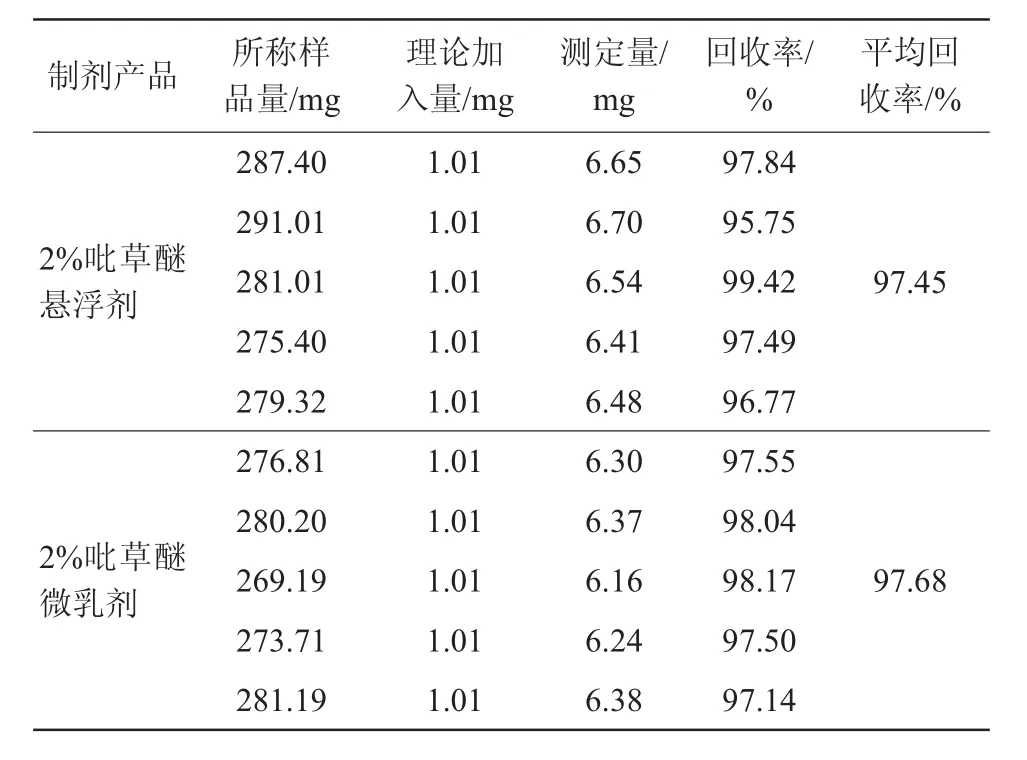
表2 分析方法的准确度试验结果
3 结 论
利用高效液相色谱法对2%吡草醚悬浮剂和2%吡草醚微乳剂分别进行了定性和定量分析。结果表明,2%吡草醚悬浮剂和2%吡草醚微乳剂的回收率分别为97.45%和97.68%。所采用的高效液相色谱法准确度和精密度较高,线性关系较好,具有简便、快速、准确及分离效果好等优点,能够作为吡草醚制剂的分析测定方法。
猜你喜欢
农药科学与管理(2022年6期)2022-08-03
新型工业化(2022年3期)2022-06-18
安徽农学通报(2022年6期)2022-04-07
北京航空航天大学学报(2021年9期)2021-11-02
天然气与石油(2021年3期)2021-07-02
建材发展导向(2021年6期)2021-06-09
中国食品(2020年16期)2020-08-31
——第二部分:原棉短纤维率标样的验证试验分析
中国纤检(2020年7期)2020-07-22
现代职业教育·高职高专(2020年30期)2020-03-16
中国航海(2019年2期)2019-07-24
