
Investigation of Salt Stress Response Mechanisms Using Mirna Sequencing of Salt Tolerant and Salt Sensitive Jute Genotypes
2020-09-09 07:31WUYupengYANGZemaoSUJianguangLIUWenxiang
WU Yu-peng , YANG Ze-mao , SU Jian-guang , LIU Wen-xiang
1. Institute of Bast Fiber Crops, Chinese Academy of Agricultural Sciences, Changsha 410205, PRC
2. College of Bioscience and Biotechnology, Hunan Agricultural University, Changsha 410205, PRC
3. Hunan Institute of Agricultural Information and Engineering, Hunan Academy of Agricultural Sciences, Changsha 410125, PRC
Abstract Salt is a major environmental stressor for crops. MicroRNAs (miRNAs) play a crucial role in plant response to salt stress. In this study, a comparative analysis of miRNAs between salt-tolerant and salt-sensitive jute subjected to high salinity were carried out. 374 known mature miRNAs were identified and 87 potential miRNA candidates from the eight sRNA libraries were predicted. For the salt-stressed versus the control, 181 miRNAs including 45 novel miRNAs were differentially expressed. More differentially expressed miRNAs were present in TC (salt tolerant) than in NY (salt sensitive). Among NY, 58 and 27 differentially expressed miRNAs were discovered in leaves and roots, respectively, whereas among TC, 45 and 113 differentially expressed miRNAs were discovered in leaves and roots, respectively. Two common differentially expressed miRNAs were discovered in both leaves and roots of the two genotypes. 10 differentially expressed miRNAs were common across both leaves and roots only in TC, including 6 upregulated, 3 downregulated and one both up and down-regulated miRNA. The results obtained in this analysis contribute to an improved understanding of abiotic-stress tolerance in jute and will be useful for breeding programs to improve jute productivity in soils with high salinity.
Key words Jute; miRNA; Salt stress; Salt-sensitive; Salt-tolerant
1. Introduction
Salt is one of the major environmental stressors restricting plant development and growth, resulting in significant yield losses worldwide[1-2]. Environmental pollution and climate change have intensified the adverse effects of salt stress through raising the salinity of the soil. Agricultural productivity and environmental conservation depend on improving plant tolerance to salt by exploring mechanisms underlying the salt stress response.
MicroRNAs (miRNAs) play a crucial role in plant salt stress response, through modulating the expression of target genes[3]. Recently, next-generation sequencing has identified numerous salt-stress-related miRNAs and their target genes, suggesting they play a major role in plant response to high salinity. ZHOU J et al.[4]reported that miR398b targeted and down-regulated a gene encoding copper/zinc superoxide dismutase (SODC) with an opposite expression pattern to miR398b. Additionally, FanmiR73 expression is reduced in salt stress, thus promoting the expression of target gene ABI5, an important TF in the ABA signaling pathway[5]. Downregulated miRNAs underlying salt stress identified in previous research include miR1029, miR164, miR165, miR168, miR319, miR444, miR5048, and miR855[3,6]; upregulated miRNAs identified in previous research include miR156, MiR395, miR171, miR393, miR6213, and miRn029[3,6-7]; up and down-regulated miRNAs identified in previous research include miR159, miRn5, miRn6, miR398, MiR394 and miR169[1,6,8-10].
Jute (Corchorus), of which there are more than one hundred species, includes two commercially cultivated species, Corchorus capsularis L. and Corchorus olitorius L., and is one of the most important crops for natural fiber production in the world[2]. Global demand for jute has recently increased because of its eco-friendly characteristics and broadspectrum application[11]. As environmental pollution and climate change have intensified the adverse effects of salt stress through raising the salinity of the soil, it is important to elucidate the salt tolerance mechanism of jute to improve its salt tolerance and expand its cultivation in saline environments. Although several studies have been conducted investigating the physiological, morphological and proteomic components of salt tolerance in jute[2], there is no available molecular research on salt tolerance in jute presently. miRNA studies on jute are limited[12-15]and are focused on miRNAs under nonsalt-stressed conditions. Hence, understanding salt stress response mechanisms in jute and improving jute salt tolerance is critical. To answer this need, in the present study, we carried out high-throughput sequencing on eight miRNAomes in leaves and roots from salt-sensitive (NY) and salt-tolerant (TC) jute genotypes. The objective was to identify potential salt stress responsive miRNAs and investigate salt stress response mechanisms in jute.
2. Materials and Methods
2.1. Plant materials and salt stress treatment
Two Corchorus olitorius L. genotypes, NY (salt stress sensitive) and TC (salt stress tolerant) were hydroponically cultivated (in Yoshida nutrient solution) at 25~28℃ in a greenhouse. Each genotype was grown in two pots containing 30 plants per pot. At the nine-leaf stage, three uniform seedlings were selected from each pot and transferred to four new pots (one control and one treatment per genotype). For each genotype, three uniform seedlings in a pot were used as the control group, while the other three uniform seedlings in the other pot were treated with 250 mmol/L NaCl Yoshida nutrient solution. After 12 h, roots and leaves from every plant in the control and treatment groups were collected for RNA extraction.
2.2. RNA isolation
Total RNA was isolated from each tissue sample using Trizol (Invitrogen, CA, US) following the manufacturer’s instructions. RNA degradation and contamination was monitored in 1% agarose gel. RNA purity was confirmed using the NanoPhotometer® spectrophotometer, and concentration was measured using the Qubit® RNA Assay Kit in Qubit® 2.0 Flurometer (Life Technologies, CA, USA). RNA integrity was assessed using the RNA Nano 6000 Assay Kit of the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA). Eight sample pools were used for miRNA sequencing (NYCR: NY control root; NYCL: NY control leaf; NYSR: NY salt-stressed root; NYSL: NY salt-stressed leaf; TCCR: TC control root; TCCL: TC control leaf; TCSR: TC salt-stressed root; TCSL: TC salt-stressed leaf). Each sample pool was composed of equivalently mixed RNA of the same tissue from the three seedlings in a pot.
2.3. Small RNA sequencing
Eight small RNA (sRNA) sequencing libraries were generated using the NEBNext® Multiplex Small RNA Library Prep Set for Illumina® (NEB, USA), following the manufacturer’s instructions. Index codes were added to attribute sequences to each sample. Library quality was assessed on the Agilent Bioanalyzer 2100 system. The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq SR Cluster Kit v3-cBot-HS (Illumina, CA, USA) for sRNA libraries following the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on an Illumina Hiseq 2500 platform. 50-bp single-end reads were generated.
2.4. sRNA analysis and miRNA identification
Raw data were quality-filtered using custom Perl and Python scripts to remove low-quality reads and those containing poly-N (poly-A, T, G, or C), 5′ adapter contaminants, or those lacking a 3′ adapter or the insert tag. All subsequent analyses were performed on high-quality clean data. The sRNA tags were mapped to reference sequences (TAIR10 genome, https://www.arabidopsis.org/) in Bowtie without mismatch, to analyze their expression and distribution on the reference sequences. Mapped sRNA tags were used to look for known miRNA, with miRBase 20.0 as a reference. The rRNA, tRNA, snRNA, and snoRNA sequences were removed using BLAST plus the Rfam and NCBI GenBank databases. Modified software miRDeep2[16]and srna-tools-cli were used to obtain the potential miRNAs and draw their secondary structures. In addition, miREvo and miRDeep2 were integrated to predict novel miRNAs through exploring the secondary structures. Furthermore, the novel miRNA sequences were blasted to the jute novel miRNA predicted by ISLAM et al.[13]. We used known miRNA and miFam.dat (http://www.mirbase.org/ftp.shtml) to identify miRNA families. Novel miRNA precursors were submitted to Rfam (http://rfam.sanger.ac.uk/search/) for matching to non-coding and structured RNA families.
2.5. Differential expression analysis of miRNA
Expression levels of miRNA were estimated by TPM (transcript per million) following previously published criteria[17]. Differential expression analysis of the treatment versus control was performed using the DESeq R package (1.8.3), and the q-value (adjusted P-value) was calculated[18]. The q-value < 0.01 and |log2(foldchange)| > 1 was set as the significance threshold for differential expression.
3. Results
3.1. High-throughput sequencing of small RNAs
We obtained 1 204 397 278 clean reads (96.68% of all raw reads) from eight small RNA (sRNA) sequencing libraries. Of the total sRNA, 62 337 791 (61.86%) reads were mapped to the reference genome (Table 1). The distribution of the number of mapped sRNA reads varied from 5 236 451 to 9 378 911 in the eight libraries. Total sRNA size distribution (18~30 nt) was nearly identical across libraries; 24-nt sRNAs were predominant, followed by 21-nt sRNAs (Fig.1).
3.2. Mature miRNA identification
We identified 374 known mature miRNAs from the eight sRNA libraries. Salt stressed TC leaves (TCSL) contained the most known mature miRNAs (296), followed by the NY leaves control (NYCL) (279), and the salt stressed TC roots (TCSR) contained the least (193) (Fig.2). In total, 340 known miRNAs were assigned to 46 defined miRNA families; thetop three included miR159 (33), miR171_1 (28), and miR166 (26).
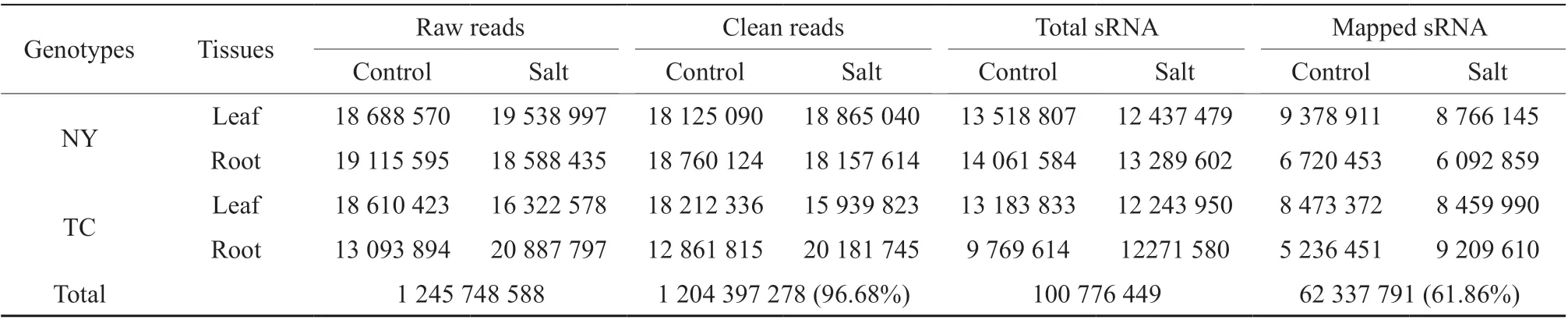
Table 1 Details of the raw and cleaned data from sequencing eight NY and TC small RNA

Fig.1 Total sRNA size distribution in eight libraries
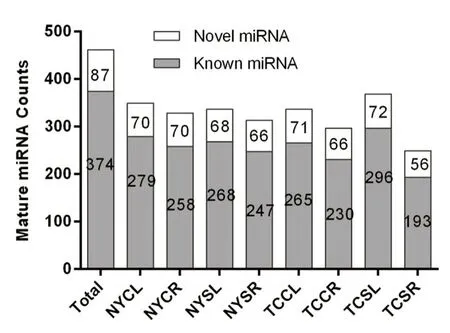
Fig.2 Statistics for known and novel mature miRNAs
We predicted 87 potential miRNA candidates (novel_1-novel_87) distributed from 56 to 71 across the eight libraries (Fig.2). Sixteen novel miRNAs were assigned to nine defined miRNA families; the miRNA families included miR171_1 (novel_56, novel_54, novel_48, novel_61 and novel_68), miR169_2 (novel_40), miR159 (novel_25, novel_3 and novel_4), miR164 (novel_51 and novel_81), miR167_1 (novel_15), miR390 (novel_46), miR394 (novel_31), miR396 (novel_23) and miR397 (novel_77).
3.3. Differential miRNA expression in response to salt stress
We identified differentially expressed miRNAs in salt stressed versus control leaves and roots from both genotypes. Overall, differential miRNA expression profiles were more similar within the same tissue type (either leaf or root), rather than between genotypes (Fig.3). For the salt stressed versus the control, 181 miRNAs including 45 novel miRNA (Table 2) were differentially expressed. Among NY, 58 miRNAs were differentially expressed between salt stressed and control leaves (NYSL vs. NYCL; 41 upregulated, 17 downregulated), whereas 27 were differentially expressed between salt stressed and control roots (NYSR vs. NYCR; 15 upregulated, 12 downregulated) (Fig.4). Among TC, 45 miRNAs in salt stressed versus control leaves (TCSL vs. TCCL) were differentially expressed (30 upregulated, 15 downregulated), while 113 miRNAs were differentially expressed in roots (TCSR vs. TCCR; 67 upregulated, 46 downregulated). 10 (5 upregulated, 0 downregulated) and 19 (14 upregulated, 3 downregulated) differentially expressed miRNAs were common across the two tissues (leaves and roots) in both NY and TC. 17 (12 upregulated, 1 downregulated) differentially expressed miRNAs were common across both NY and TC leaves, while 11 (4 upregulated, 2 downregulated) were common across both NY and TC roots. Two miRNAs (stu-miR398a-3p and zma-miR398a-3p) were upregulated in both TC leaves and roots, and upregulated in NY leaves but downregulated in NY roots. 10 differentially expressed miRNAs were common across both leaves and roots only in TC, including 6 upregulated miRNA (aau-miR168, gma-miR172b-5p, gma-miR396b-3p, mes-miR172c, ptc-miR396e-3p, and vvi-miR172a), 3 downregulated miRNA (ath-miR396b-5p, bdimiR159a-3p, and vvi-miR396b) and one (osamiR397b) that was both up and down-regulated, suggesting possible mechanisms uniquely related to salt tolerance.

Table 2 Differentially expressed novel miRNA in multiple comparison groups.
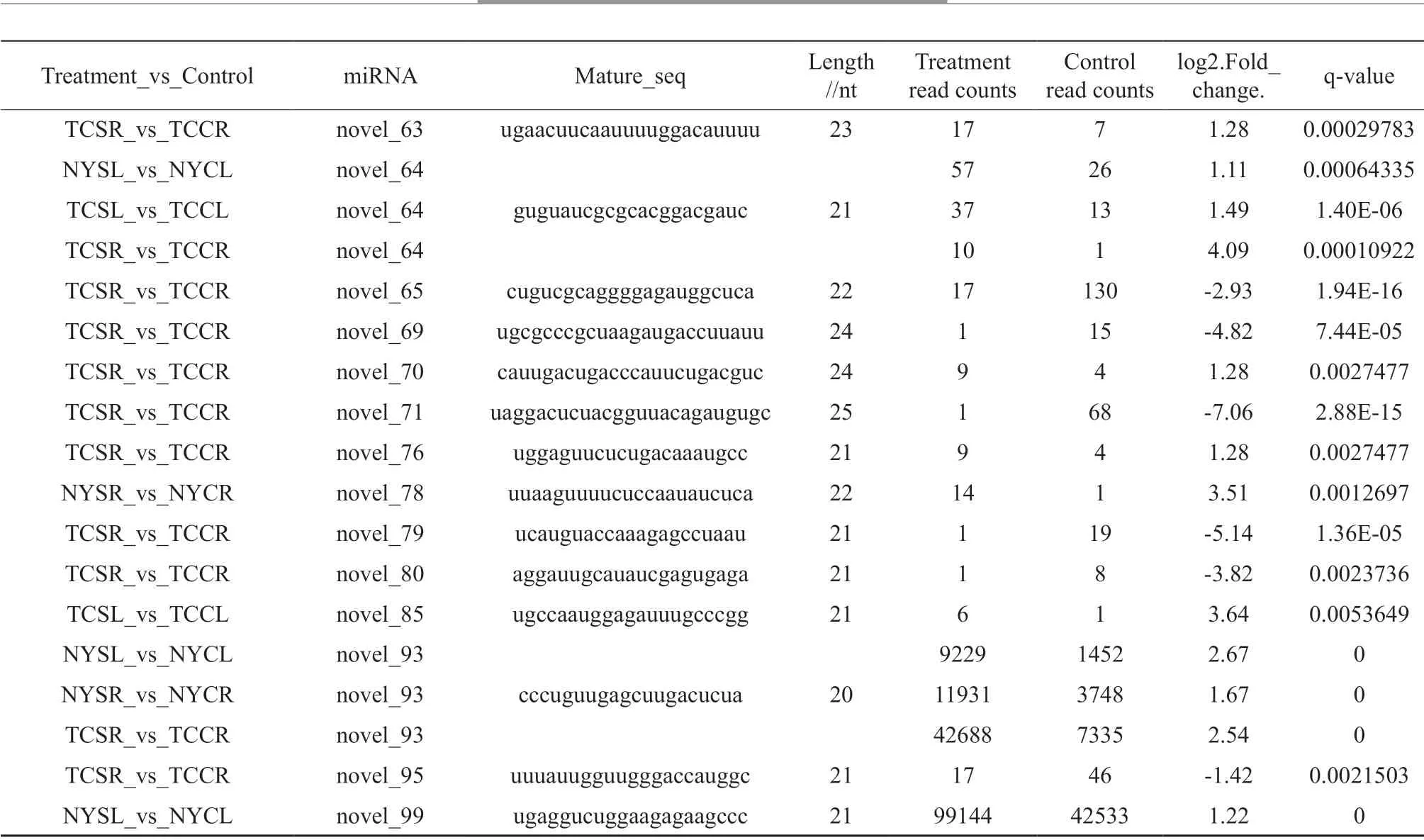
NYCR: NY control root; NYSL: NY salt-stressed leaf; NYCL: NY control leaf; TCSR: TC salt-stressed root; TCCR: TC control root; TCSL: TC saltstressed leaf; TCCL: TC control leaf.
4. Discussion
MicroRNAs (miRNAs) play a crucial role in plant responses to salt stress and are a potential genetic approach for understanding stress tolerance at the molecular level[19]. Recently, next-generation (highthroughput) miRNA sequencing has greatly increased the effectiveness of mining for stress-resistance-related miRNAs that explain the stress response mechanism in plants. Some stress-resistance-related miRNAs in plants have been identified, including the most wellknown salt stress responsive miRNAs (miR396, miR172, miR168, miR169, miR159 and miR156)[20-22]. There is currently a few miRNA studies on jute[13,15,23], whereas there has been no miRNA research on jute’s salt-stress response mechanisms. In this study, we performed miRNA sequencing to explore the salt tolerance mechanisms in the leaves and roots of two jute genotypes (salt resistant (NY) and salt sensitive (TC)) under salt stress. We identified 374 known mature miRNAs and predicted 87 novel mature miRNAs, more than previous reports on jute[15]. The sRNA size distribution (18~30 nt; predominantly 24-nt, then 21-nt) was generally similar to findings from previous studies in rapeseed, maize, and Sedum alfredii[24-25], differing only slightly from a report with sRNAs predominantly at 21-nt, followed by 24-nt[13].
In our study, 181 miRNAs were differentially expressed under salt stress. More differentially expressed miRNAs were present in TC than in NY. Previous studies[5]have shown that in plants, gene-expression regulation occured at the posttranscriptional level via miRNAs under stress conditions. This implied that the tolerant genotype may exhibit more post-transcriptional regulation that contributed to its higher resistance under salt stress. We found 10 differentially expressed miRNAs from five miRNA families (one miR168, three miR172, four miR396, one miR159, and one miR397) only in salt stressed TC. These miRNA families were all related to plant stress resistance[26]. For example, miR168 regulated ARGONAUTE1 (AGO1) homeostasis, post-transcriptional control of which might play an important and conserved role in the stress response[27]. And previous studies have reported the miRNAs represented differential expression after salt shock in Arabidopsis[28], Caragana[29]and Zea mays[30]. It is likely that these ten miRNAs play a role in TC salt tolerance being similarly important to that it plays in other plant species[31-33].
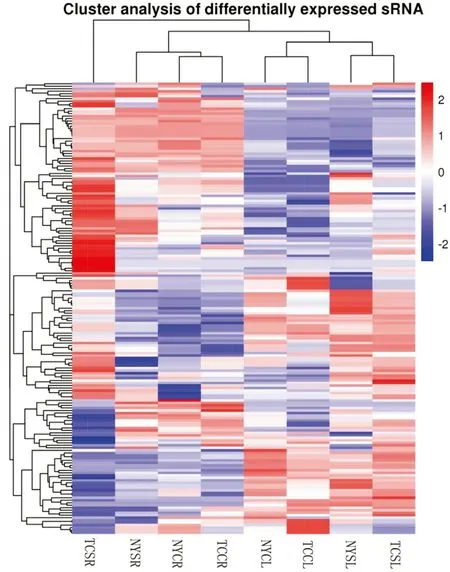
Fig.3 Cluster analysis of differential miRNA expression profiles
The miR398 was considered a bridge linking plant responses to oxidative stress and other stresses such as salt stress and water deficit[34]. miR398 targeted CSD1 encoding superoxide dismutase scavenging excess reactive oxygen species (ROS) in plants exposed to salt stress[35]. However, transcriptional regulation showed a different pattern; two differentially expressed miR398 family members associated with plant salt tolerance were common across leaves and roots from both genotypes. The miRNA stu-miR398a-3p was upregulated in leaves and roots from both genotypes, whereas zmamiR398a-3p was upregulated in TC leaves and roots, and upregulated in NY leaves and downregulated in NY roots. These results suggested dynamic miR398 regulation across different tissues and genotypes, being consistent with previous reports[36]. Downregulated miR398 in NY roots might be beneficial for increasing superoxide dismutase and scavenging excess reactive oxygen species (ROS) under salt stress.
Forty-five of 87 novel miRNA presented differential expression. These miRNAs were not reported in Islam’s study[13], possibly because Islam’s study was on one treatment environment and the sample pool was seedlings, but our study was of two treatment environments (salt stress and control) and two tissues (leaf and root). Some novel miRNAs were discovered in more than one treatment group. For example, novel_5 and novel_64 were upregulated across both leaves and roots in TC and in NY leaves; novel_20 and novel_37 were upregulated across both leaves and roots in NY; and novel_25 and novel_40 were downregulated in roots and leaves, respectively, of both genotypes. The differentially expressed novel miRNA was most likely important for jute salt tolerance. The five differentially expressed novel miRNA were new members of miR396 (novel_23), miR159 (novel_25), miR169_2 (novel_40) and miR171_1 (novel_56 and novel_61), these families were important in responding to salt stress. As seen from the above, the mechanism of miRNA response to salt stress needed further study.
In summary, our study provided a comparative analysis of miRNAs between salt tolerant and salt sensitive jute subjected to high salinity. miRNA analyses were still extremely rare in jute, despite the fact that the crop was very important. The results obtained in this analysis contributed to an improved understanding of abiotic-stress tolerance in jute. Moreover, our data on miRNA should prove useful for breeding programs to improve jute productivity under the condition of soil salinity.
 Agricultural Science & Technology2020年2期
Agricultural Science & Technology2020年2期
- Agricultural Science & Technology的其它文章
- Effects of Nitrogen Fertilizer on Grain Quality of Different Cultivars of Japonica Rice
- Analysis on Some Agronomic Characters and Pedigree in Hezihao Series Peanut Varieties
- A Study on Planting Adaptability of Various Soybean Varieties in Hengyang
- Analysis of Rhizosphere Microbial Community Structure in Different Pathogenesis Stages of Watermelon Fusarium Wilt
- Study on Extraction Method of Carotenoids from Citrus Leaves
- Optimization of Steam Explosion Process Condition for Extracting Polysaccharides from Pseudostellaria heterophylla by Response Surface Methodology