
1个北村网状肢端色素沉着症大家系ADAM10基因突变研究
2020-09-09 08:25:56葛宏松曹婷婷吴建平
安徽医科大学学报 2020年8期
葛宏松,张 成,周 洁,曹婷婷,董 瑛,吴建平,冉 颖,张 莉,李 明
北村网状肢端色素沉着症(reticulate acropigmentation of Kitamura,RAK)是一种罕见的色素障碍性遗传疾病,常染色体显性遗传,外显率高[1],1943年由 Kitamura和Akamatsu首次报道后,在日本、印度、中国、尼泊尔、土耳其、伊朗、沙特阿拉伯、意大利和拉丁美洲等国家和地区人群中均有单个病例及家系报道[2-4]。但该病与屈侧网状色素异常病(dowling-degos disease,DDD)等[1]色素障碍性疾病在临床上有许多相似性,过去由于技术原因,只能根据这些疾病的分布、形态、排列、发病年龄和有无低色素斑点对各种实体进行描述,同时该病本身也存在较大的临床异质性,往往造成很多误诊。随着分子遗传学技术发展,一系列单基因色素障碍性疾病的致病基因被发现,这些疾病的诊断才逐渐清晰。2013年,Kono et al[5]首次确定了RAK的致病基因ADAM10,从而从基因水平区分开与其它临床表现相近的色素障碍性疾病。该研究报道了在中国人群RAK临床特点及新的ADAM10 基因突变位点。
1 材料与方法
1.1 家系成员调查先证者(46号患者):女性,27岁,其女(53号患者)患有湿疹来安徽医科大学附属省儿童医院皮肤科就诊,发现其家族成员有多名患者,通过先证者联系对家系进行现场调查,制作调查表以了解家族中各成员的临床表现及发病情况,并绘制家系图。
1.2 血细胞基因组DNA提取及分离经过患者及其他正常人签署遗传性皮肤病研究知情同意书后,采集家系中外周血样本(患者血样10例,正常对照12例),同时采集无血缘关系中国汉族正常对照血样100例,琼脂糖凝胶电泳检测DNA的完整性和数量。使用血液DNA试剂盒[6](美国普罗米加公司)分离出DNA,该研究通过安徽医科大学附属省儿童医院伦理委员会审查,符合赫尔辛基宣言。
1.3 PCR扩增及基因测序在线(http://www.genome.UCSC.edu/)查取POFUT1、POGLUT1、ADAM10、KRT5 以及 ADAR1外显子及其剪接区域进行检测,根据公布的基因序列设计ADAM10基因全部编码外显子的特异性引物(表1)进行PCR扩增,方法如下:5 min内升温至94 ℃后,持续变性(94 ℃、30 s),退火(55~60 ℃、30 s),延伸(72 ℃、45 s),共35个循环,最后充分延伸10 min。应用2%琼脂糖凝胶电泳检测提取的PCR产物。PCR产物送上海安百隆生物技术服务有限公司进行正向测序,测序结果与以上5种人类基因组序列比较,如发现突变再行反向测序进行验证。
2 结果
2.1 临床表型资料共发现该家系5代共有成员54人(其中患者16人,正常38人,男25人,女29人,11人已去世),家系中男女均发病(其中男患者7人,3人去世,女患者9人,3人已去世),无隔代遗传现象,符合常染色体显性遗传模式(图1)。家系中先证者及所有患者发病年龄在10岁以后,皮疹逐渐加重,查体:一般情况可,颈部及手部的网状色素沉着斑,未见色素脱失斑,无掌跖部凹陷损害,无其它系统性病变发现,先证者皮损组织病理显示,色素斑处表皮轻微萎缩,角质形成细胞内黑素蓄积,基底层色素细胞增多(图2)。

表1 扩增ADAM10基因16个外显子引物序列及基因片段长度
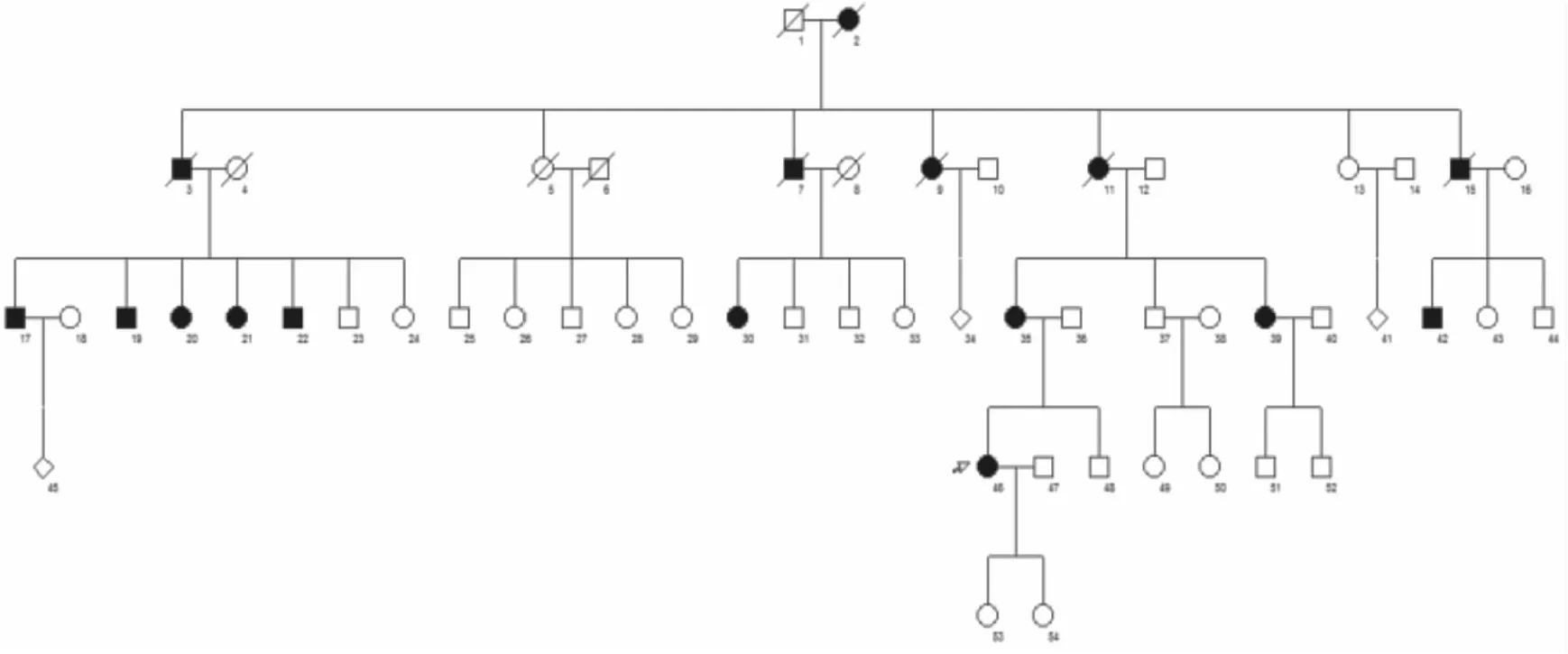
图1 RAK家系分布图
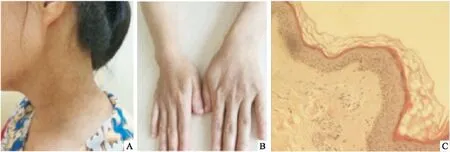
图2 RAK家系中先证者

图3 RAK 家系患者ADAM10基因突变检测
2.2 基因检测结果每个样品提取得到2 μg以上的基因组DNA,用琼脂糖凝胶电泳检测显示均无明显拖尾,基因组DNA完整性较好。各PCR产物用琼脂糖凝胶电泳检测显示条带单一,片段大小符合预期。基因测序结果与参考序列比对显示均为特异性扩增。引起色素异常的POFUT1、POGLUT1、KRT5 以及 ADAR1 基因未发现致病突变,而在ADAM10基因的第 4 号外显子发生移码突变,即 c.425-426 delGA; p.R142IfsX2,该突变在正常对照家系成员及无血缘关系的 100 例正常对照中并未检测到,突变导致该蛋白截短,引起单倍型剂量不足,从而导致疾病的发生(图3)。
3 讨论
RAK的特征性表现为色素沉着、角状、不规则的斑点样病变,表面萎缩,在手、足背、颈部呈网状排列[1]。病变通常开始于儿童期或青春期,逐渐扩展到四肢,很少发生在面部和眼睑。随着时间的推移,病变逐渐变暗,阳光会使病情恶化。掌心、脚底和指骨背表面的皮纹有凹坑和裂口,伴掌跖皮肤纹理破坏。色素斑的病理检查可见表皮萎缩,表皮突延长,黑素增加。但不是所有患者均具有掌跖和指(趾)表面小凹陷,皮肤纹理破坏。本组患者表现为网状的雀斑样色素沉着斑分布于手足背面及颈部,皮疹表现为萎缩性的斑片,无色素减退斑及掌跖部虫蚀样损害,所有患者大约10~20岁间发病,逐渐加重,未见其它系统性损害。
RAK的鉴别诊断以网状或点状色素沉着为表现的疾病,包括网状色素性皮病[1](dermatopathia pigmentosa reticularis,DPR):2岁左右婴儿期发病,躯干部多见,可累及颈、肩、大腿等部位,伴有色素沉着、非瘢痕性秃发和甲病三联征,组织病理为真皮上部大量色素及噬色素细胞。遗传性泛发性色素异常症(dyschromatosis universalis hereditaria,DUH):大多数1岁内发病,晚发偶见,全身皮肤甚至黏膜可见界限清楚的棕色及白色斑,混杂分布,主要累及躯干及四肢近端,偶伴系统损害,组织病理示表皮色素颗粒增加及色素失禁,Xing et al[7]将此病的致病基因定位于6号染色体6q24.2-1q25.2区域。遗传性对称性色素异常症(dyschromatosis symmetrica hereditaria,DSH):一种肢端色素沉着病,通常起于婴儿期或幼儿期,青春期明显,持续终身,表现为手、足背点状黄褐色至褐色斑点夹杂脱色素斑疹,构成网格样图,组织病理表皮下层色素增加真皮上层噬色素细胞多见及少量炎性淋巴细胞,脱色素斑处基底层色素减少甚至消失。2003年Zhang[8]课题组首次将DSH致病基因定位于1号染色体 1q11-1q21区域内,随后发现致病基因ADAR[9]。家族性进行性色素沉着症褐色斑出生后发生,随年龄增大逐渐增多,好发于眼周、口周、颈侧、躯干、四肢及手足背部雀斑样斑点,其间可见正常皮肤,病理示基底层色素增加、色素细胞正常,Wang et al[10]在国际上首先明确该病的致病基因为KITLG。DDD一般为20岁以后发病,主要见于颈、乳房下、腋窝、臀间、腹股沟等屈侧花纹样棕褐色斑,也可累及躯干和前臂, 近年来,关于RAK与DDD共存于同一患者的报道越来越多,但在2006年,Betz et al[11]首先发现部分DDD是由KRT5基因突变所致;在2013年[12],排除了KRT5基因突变后,国际上首次报道了泛发性DDD是由POFUT1基因突变所致,Kono et al[5]首次应用全外显子组测序确定ADAM10突变是造成RAK的原因,这是一种不同于DDD等其他色素POFUT异常性疾病,同时,在5个RAK家系中,确定了6种ADAM10基因突变。在RAK家族中发现了ADAM10基因突变,该基因编码锌金属蛋白酶、崩解素和ADAM 10。众所周知,ADAM 10参与了皮肤中各种基质的外域脱落,因此,遗传研究显示RAK和DDD是不同的疾病。2018年,Pan et al[13]在2例单独发病的中国RAK患者中,通过测序分析检测出ADAM10内含子突变(c.2026-2A >G)。ADAM10蛋白参与多种生物过程,包括调节角质形成细胞中黑色素的分布和转运过程。迄今为止,包括本组患者在内,共报道了14个ADAM10突变导致RAK。在这些患者中,甚至在ADAM10突变相同的家庭成员中,也发现了色素沉着和凹陷分布的多样性,以及其他特征的存在,如特应性皮炎、脑膜瘤和足底角化病。RAK的表型复杂性提示,除了ADAM10突变外,其他迄今未知的环境或遗传因素也可能与RAK的发病机制有关。
1998年,Yavari et al[14]通过FISH技术把ADAM10基因定位于15q22。人类ADAM10的mRNA含3 410个碱基,16个外显子,基因的上游区域可能存在BRN2、SREBP、OCT1和NFKB等的结合位点。Hartmann et al[15]建立了ADAM10缺陷小鼠,该小鼠在胚胎9.5 d死亡,通过原位杂交显示,该小鼠NOTCH 靶基因 Hes5在神经管中的表达下降,而NOTCH配体DLL1的表达则增加,表明 ADAM10在NOTCH信号通路中发挥着重要作用。进一步的研究还显示,ADAM10 是小鼠边缘区B细胞发育必不可少的。缺乏ADAM10的B细胞 NOTCH2信号通路严重受损,ADAM10作为CD23的释放酶,可能以某种不可被替代的机制,启动了NOTCH2信号通路。Kono et al[5]报道ADAM10单倍体功能不足引起无毛小鼠皮肤上雀斑样色素沉着,小鼠的色素斑首发于四肢背侧,成年鼠躯干可观察到弥漫性色素沉着。这一表现类似于人类的RAK。但两者明显的区别是,小鼠出现色素沉着需要同时具备ADAM10突变致单倍体功能不足和无毛基因纯合突变两个条件。而人类RAK患者,全身毛发正常,且经检测,也不存在无毛基因的任何突变。
综上,虽然以前的研究已经明确RAK是由ADAM10基因异常所致,也已经建了两个ADAM10突变小鼠模型,但这两个模型一个在胚胎期即死亡,另一个需同时合并无毛基因的纯合突变,均与人类RAK有明显的不同,不适合进行 RAK发病机制的研究。所以,ADAM10基因突变如何影响人类的黑素形成,如何导致人类皮肤的色素代谢障碍,最终使患者的皮肤表现出RAK 表型,需要进一步研究。本研究中患者的临床仅表现为颈、手部的网状色素沉着斑,无掌跖部凹陷损害,新ADAM10基因4号外显子突变的发现,丰富了该病突变库,同时对该家族成员今后的产前诊断、优生优育有一定的帮助。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29 01:57:52
世界科学技术-中医药现代化(2022年2期)2022-05-25 13:18:28
昆钢科技(2022年1期)2022-04-19 11:36:16
世界科学技术-中医药现代化(2021年12期)2021-04-19 12:31:20
中国生殖健康(2020年4期)2021-01-18 02:58:10
中国生殖健康(2018年4期)2018-11-06 07:12:16
广东海洋大学学报(2015年4期)2016-01-13 08:39:30
听力学及言语疾病杂志(2015年5期)2015-12-24 01:47:04
首都医科大学学报(2015年4期)2015-12-16 13:00:08
郑州大学学报(医学版)(2015年2期)2015-02-27 14:50:44
