
高通量测序多基因检测对家族性心房颤动的预测价值∗
2020-09-02 07:10:02刘俊陈舒应璇叶晓霞董扬王文标
中国心脏起搏与心电生理杂志 2020年4期
刘俊 陈舒 应璇 叶晓霞 董扬 王文标
心房颤动(简称房颤)是最常见的心律失常,其中呈现明显家族性者称为家族性房颤(familial atrial fibrillation,FAF)。房颤电生理机制和病理生理机制复杂,发病机制尚未完全清楚。目前,越来越多的学者将研究的目光投放在遗传因素上[1-2]。据流行病学研究报道,30%的房颤患者具有房颤家族史[3],5%的典型房颤患者以及15%的孤立性房颤患者呈现明显家族遗传性[4],说明FAF患者数目可能比预想中的要多。与此同时,学者们从庞大的基因组中探寻到数种可能与房颤相关的基因变异[5],然而尚少见关于FAF相关致病基因的研究。这些遗传变异影响了离子通道、电传导、信号传导通路等生理机制,最终破坏正常电兴奋[6-7]。因此,对FAF致病基因的研究不仅是为明确突变位点,还能探索遗传因素导致房颤发生的分子学机制,为挖掘其他类型房颤发生机制提供新的思路与理论依据。本研究基于此,笔者对比已知房颤 相 关 基 因KCNQ1、KCNE2、SCN5A、KCND3、CJA5、ACC9在FAF、非FAF患者以及正常无房颤者间的检出率,研究上述致病基因与FAF相关性及其对后者的预测价值。
1 资料与方法
1.1 研究对象及分组
应用随机数字表法选取2017年1月至2018年10月间于金华市人民医院就诊的40例FAF 患者作为FAF组,并选取20例同期非家族性房颤患者纳入非FAF组,以及纳入20例正常无房颤者作对照组。纳入标准[8]:①FAF 组具有明显家族史,遗传模式符合孟德尔遗传模式,三代亲属中≥3例发病,患者心电图、动态心电图或植入式起搏器等监测到房颤心电图;②年龄:20~50岁;③已签署知情同意书;④非FAF组和对照组患者三代亲属中均无遗传性心律失常及遗传性心脏离子通道病,非FAF组有明确房颤病因(如甲亢、高血压性心脏病、扩张型心肌病等)。排除标准:①半年内存在心肌梗死病史;②近1月内接受心脏手术者;③纽约心脏病协会(NYHA)分级3、4级心力衰竭者;④存在严重心脏瓣膜病变、甲状腺功能减退、水电解质紊乱、严重肺功能障碍者;本实验设计已由医院伦理学委员会审核。
1.2 方法
所有入组研究对象于门诊或入院后24 h内被收集病史、既往史、年龄、身体质量指数(BMI)、心电图结果等基本信息。
1.2.1 基因组DNA 的提取 采集所有入组人员外周抗凝血6 ml,应用QIAamp DNA Blood Mini Kit(NO.51106)试 剂 盒(购 自 德 国QIAGEN 公司)对样本进行基因组DNA 提取。DNA 质量浓度≥50 ng/μL,吸光度(A)260/A280为1.8~2.0,DNA 总量≥6μg。
1.2.2 目标基因测序 根据NCBI数据库获取致病基因(KCNQ1、KCNE2、SCN5A、KCND3、CJA5、ACC9)所对应的序列(具体交由杭州本因生物科技有限公司完成),使用安捷伦SureSelect靶向序列捕获系统设计特异性寡核苷酸链探针,其中包含目标基因所有外显子区域、外显子和内含子交界区域以及全部基因的5′和3′末端。将1μg基因组DNA样本打断成长为200~500 bp的片段,末端补平、加“A”,与寡核苷酸混合物接头衔接,将所得产物进行PCR 反应扩增、纯化,构建DNA 文库并对其行质量检测。预先设计的特异性捕获探针与合格的DNA文库杂交、富集后,将富集的外显子区域的DNA 片段洗脱下来,并检测富集效率。然后,应用第二代高通量测序技术将捕获的DNA 文库在Illumina HiSeq TM2000平台(美国Illumina公司提供)上对目标序列进行测序,测序深度200X,得到原始数据并确保所有检测样品均达到预期测序标准,Q30平均比例在80%,平均错误率在0.1%以下。探针设计标准、捕获条件、基因组DNA 与捕获探针比例参考文献[9]。
1.2.3 生物学信息分析 应用Illumina basecalling Software 1.7 软件对原始测序数据进行处理和控制,并将其与NCBI人类基因组DNA(GRCh37/hg19)参照序列对比。应用GATK、AtlasSNP、Atlaslndel2软件分析并获得单核苷酸多态性、小插入与小缺失变异相关信息,了解样本中的DNA 序列变异。将检测获得的DNA 变异序列信息与1000 Genome、dbSNPI 35、NHLBIexome sequencing database、NIEHS exome sequencing database数据库进行对比,去除高于0.5%的隐形突变、高于0.1%的显性突变、对蛋白功能无影响的突变,最终得到FAF相关突变位点。本研究所纳入的KCNQ1、KCNE2、SCN5A 等致病基因中,若检测出错义突变或有意义的碱基突变则计作阳性(+),由此计算致病基因突变检出率。
1.3 统计学方法
采用SPSS 25.0 版统计学软件分析,年龄、体重、BMI等采用(±s)表示,组间比较采用两独立样本t检验,三组间比较采用方差分析,两两比较采用bonferroni校正。计数资料用例数和百分数表示,采用χ2检验,应用多因素Logistic回归分析探究致病因子与FAF 相关性,以P<0.05 为差异有显著性。
2 结果
2.1 三组一般临床资料比较
三组年龄、性别、体重、BMI等资料比较均无明显差异(P>0.05),见表1。
2.2 三组致病基因突变检测结果比较
三组致病基因KCNQ1、KCNE2、SCN5A 突变检出率存在统计学意义(P <0.05),而KCND3、CJA5、ACC9基因突变检出率无明显差异(P >0.05),见表2。

表2 三组致病基因突变检测结果比较[(n)%]
2.3 致病基因突变与FAF相关性比较
根据三组患者致病基因检出率结果,选择其中P<0.05的3个可能与FAF 相关的致病基因进行Logistic回归分析。分析显示,致病基因KCNQ1、SCN5A、KCNE2突变均为FAF 的独立危险因素,见表3。
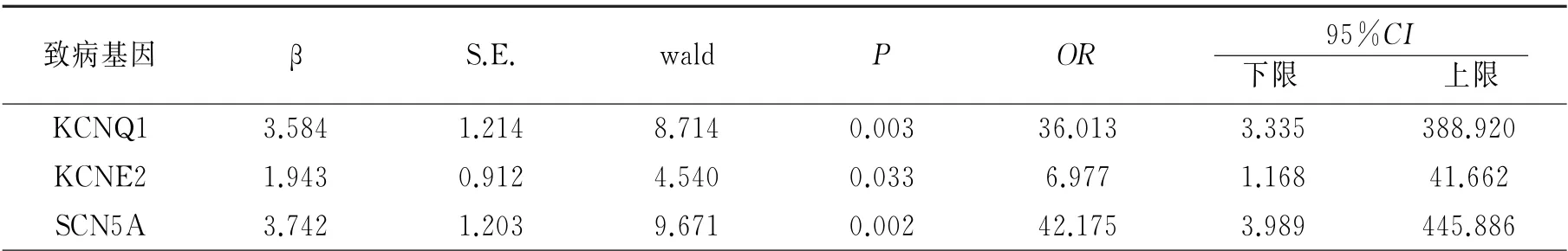
表3 FAF致病基因Logistic回归结果
2.4 致病基因KCNQ1(+)、KCNE2(+)、SCN5A(+)单独诊断FAF的诊断效能比较
不同致病基因单独检测的诊断效能比较均无明显差异(P>0.05),见表4。
2.5 不同致病基因突变联合检测方式对FAF的诊断效能比较
KCNQ1(+)、KCNE2(+)、SCN5A(+)三者两两联合检测与三者联合检测的诊断效能比较无明显差异(P>0.05),见表5。
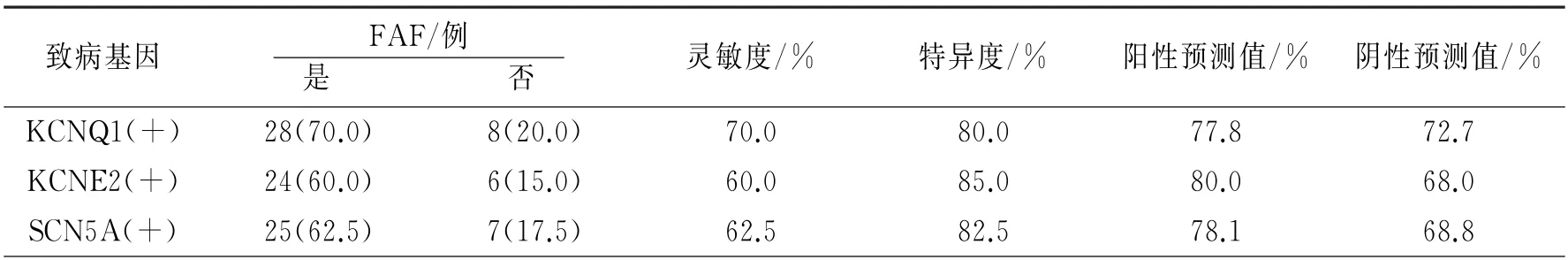
表4 不同致病基因单独诊断FAF的诊断结果比较
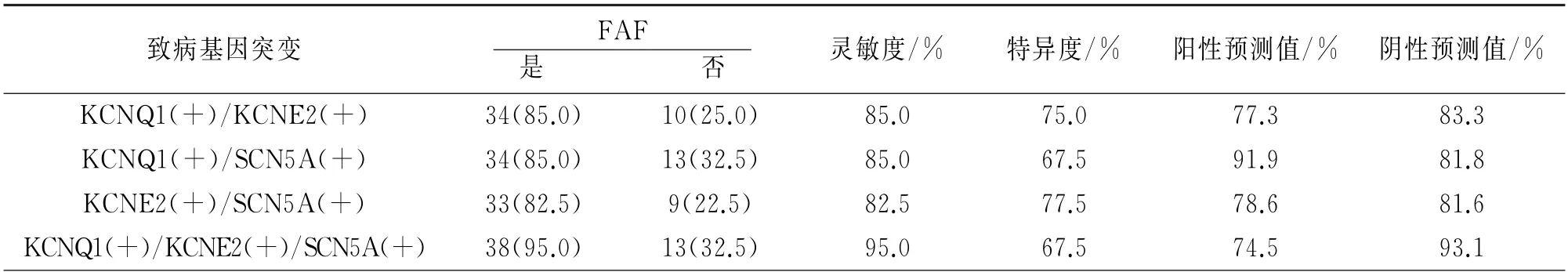
表5不同致病基因联合检测的诊断结果比较
3 讨论
随着分子生物学的不断发展,越来越多的疾病被证实与基因突变相关,自1997年Brugada首次发现房颤相关染色体位点以来,目前至少17个房颤致病基因已被人们发现[10]。但有关FAF的相关研究局限在白种人群,在亚洲人群中的研究甚少。为丰富这一领域的研究,笔者收集了40例FAF 患者的基因组DNA,将之于非FAF患者、正常群体进行对比,发现:三组致病基因KCNQ1、KCNE2、SCN5A突变检出率存在统计学意义;致病基因KCNQ1、SCN5A、KCNE2突变均为FAF 的独立危险因素,说明KCNQ1、KCNE2、SCN5A 与FAF的发生密切相关。姜永日等[4]报道:KCNQ1 基因编码的KCNQ1亚基可与KCNE1亚基结合形成通道复合体,构建成的缓慢激活延迟整流钾通道IKS在动作电位时发生失活,导致房颤发生;KCNE2可影响KCNQ1-KCNE2通道功能,缩短心肌细胞动作电位间期,从而启动房颤。Ilkhanoff等[11]研究中发现,SCN5A 基因H558R 位点多态性与房颤的发生、发展有着紧密的联系,SCN5A 基因变异rs7629265可使非洲裔美国人房颤风险提升74%,并与PR 间期的缩短有关。本实验中,笔者采用新一代高通量测序技术对研究对象DNA 文库进行测序,相比传统第一代Sanger测序技术,前者具有检测速度快、准确性高、覆盖度广等特点[12]。通过实验,笔者认为致病基因KCNQ1、KCNE2、SCN5A 可能与FAF发生具有一定相关性,这一结论可能给往后FAF相关基因多态性的研究提供理论依据。而本实验样本量较少,地域局限,未对比汉族与其他民族的实验结果差异,有待今后进一步的扩展研究。
挖掘FAF相关基因突变,可为从遗传学和分子生物学上揭示房颤致病机制带来一定的线索,同时也是为实现FAF基因诊断、研发特异性治疗方法带来新的希望。越来越的研究表明房颤发病与多个基因的多态性有关,在本实验中,致病基因KCNQ1、KCNE2、SCN5A 突变情况单独诊断FAF的诊断效能较低,不同致病基因突变情况单独检测的诊断效能比较均无明显差异,因此单独致病基因检测的诊断效能可能并不理想。本文中KCNQ1(+)与KCNE2(+)和SCN5A(+)联合检测时灵敏度和特异度分别为95.0%、67.5%,相对单独检测和两两联合检测,三者联合检测可取得较高的灵敏度,但两两联合检测与三者联合检测的诊断效能比较并无明显差异。Olesen等[13]报道单基因突变在孤立性房颤和FAF中虽然存在,但是较为罕见。单个基因突变所致的房颤只占全部房颤患者中的极少部分,更多的FAF及普通房颤患者的发病原因可能是由多个基因突变或基因-环境相互作用引起的。因此,为提高基因检测对FAF的诊断效能,我们建议将KCNQ1(+)、KCNE2(+)、SCN5A(+)三者两两联合检测或三者联合检测以获得更理想的诊断灵敏度,这不仅有助于提升基因检测对FAF的预测价值,提早对患者进行健康干预,同时也有助于发现更多的FAF患者。然而本实验样本较少,至于诊断效能最理想的致病基因联合检测方式还有待进一步的研究。
猜你喜欢
英语世界(2023年6期)2023-06-30 06:29:10
保健医苑(2023年2期)2023-03-15 09:02:50
今日农业(2021年11期)2021-08-13 08:53:24
中国生殖健康(2020年2期)2021-01-18 02:51:26
小学生导刊(2018年13期)2018-06-29 03:49:00
大众健康(2017年8期)2017-08-23 21:18:22
老友(2017年7期)2017-08-22 02:36:30
遗传(2014年3期)2014-02-28 20:58:49
世界科学(2014年8期)2014-02-28 14:58:31
河南医学研究(2014年5期)2014-02-27 14:52:41
