
生物基超韧聚乳酸复合材料的制备与性能
2020-08-26 03:11:58夏艺玮王光鑫冯玉林胡跃鑫赵桂艳
高等学校化学学报 2020年8期
夏艺玮,王光鑫,冯玉林,胡跃鑫,赵桂艳
(辽宁石油化工大学石油化工学院,抚顺113001)
聚乳酸(PLA)是一种来源于可再生资源的生物降解高分子材料,具有良好的力学性能、透明性、化学稳定性及生物相容性等[1~5],被认为是最有应用前景的生物降解高分子材料.但PLA柔韧性及耐热性较差,尤其是抗冲击性能差,限制了其应用[6~13].目前,提高PLA韧性的方法有共聚及共混等方法.其中,将PLA与弹性体或橡胶熔融共混是提高PLA韧性的经济而有效的方法,但大部分增韧剂为石油基高分子材料[14~21],无法维持聚乳酸的生物基本质.近年来,越来越多的生物基和生物降解高分子材料被用于PLA增韧改性,如聚(丁二酸丁二醇酯)(PBS),聚[(丁二酸丁二酯)-共己二酸酯](PBSA)和聚己二酸对苯二甲酸丁二酯(PBAT)[22~24]等.大部分PLA共混材料的拉伸韧性有所提高,但冲击韧性的提高非常有限.
本文以来源于可再生资源的聚醚酰胺弹性体(PEBA)为增韧剂,在共接枝单体乙烯基吡咯烷酮(NVP)的存在下,通过熔融接枝反应将极性单体甲基丙烯酸缩水甘油酯(GMA)接枝到PEBA 上,得到PEBA-GMA,然后与PLA熔融共混制备全生物基超韧聚乳酸(PLA/PEBA-GMA)复合材料,研究了接枝单体组分GMA,NVP和DCP的比例和用量对PLA/PEBA-GMA复合材料结构与性能的影响.
1 实验部分
1.1 试剂与仪器
聚乳酸(PLA),牌号4032D,美国Nature Works 公司,密度为1.24 g/cm3,=1.7×106,多分散系数为1.43;生物基聚醚酰胺弹性体,牌号PebaxRnew25R53,法国阿科玛公司,由PA11(硬段)和聚醚(软段)构成的嵌段共聚物;甲基丙烯酸缩水甘油酯(GMA),分析纯,苏州安利化工厂有限公司;过氧化二异丙苯(DCP)和正丁醇,分析纯,北京化工厂;乙烯基吡咯烷酮(NVP,纯度99%),美国Aldrich 公司;PLA和PEBA使用前需于60oC真空干燥24 h.
XSS-300型转矩流变仪,上海科创橡塑机械设备有限公司;XJU-5.5J型悬臂梁冲击试验机,承德大华试验机有限公司;Instron Model5966型静态拉伸试验机,美特斯工业系统(中国)有限公司;Q20型差示扫描量热(DSC)仪,美国TA 公司;SU-8010 型场发射电子扫描显微镜(SEM),日本日立电子株式会社;DHR-2 型流变仪,美国TA 公司;Bruker Vertex 70 型傅里叶变换红外光谱(FITR)仪,德国Bruker公司.
1.2 材料的制备
1.2.1 PEBA-GMA的制备 将50 g PEBA在180 ℃,转速为80 r/min的条件下预混1 min使其完全熔融,然后加入不同质量(1.05,2.1,3.15,5.25 g)的GMA,NVP和DCP的混合物(三者质量比均为1∶1∶0.1)继续共混5 min,制得PEBA-GMA.
1.2.2 PLA/PEBA 和 PLA/PEBA-GMA 复合材料的制备 分别将 PLA 与 PEBA 和 PEBA-GMA 按 100:0,90:10,80:20,70:30 的质量比在180 ℃,转速为80 r/min 的条件下熔融共混5 min,制得PLA/PEBA 和PLA/PEBA-GMA复合材料.
1.3 性能表征
冲击和拉伸测试样条均在180 ℃,10 MPa 条件下熔融热压成型,尺寸分别为63.5 mm×12.7 mm×3.2 mm和50.0 mm×4.0 mm×1.0 mm(颈部宽度4.0 mm).根据ASTM D-256标准,在室温下测试冲击性能,测试5次,取平均值.拉伸测试根据ASTM D-638标准,以20 mm/min的拉伸速率测试5次,取平均值.以20 ℃/min 的速率从室温升至200 ℃,恒温3 min 消除热历史;然后以10 ℃/min 的降温速率从200 ℃降至0 ℃;再以10 ℃/min的速率升温至200 ℃.根据二次升温曲线,通过下式计算PLA组分的结晶度(Xc,%):

式中:ΔHm(J/g)为熔融焓;ΔHc(J/g)为加热过程中的冷结晶焓;wf(%)为复合材料中PLA 的质量分数;为PLA完全结晶时熔融焓的理论值(93.7 J/g)[25].
以20 ℃/min 的速率从室温升至200 ℃,恒温3 min 消除热历史;然后快速降温至100 ℃进行等温结晶,直至结晶完全.通过Avrami 方程进行处理得到1-Xt= exp(-Zttn), 式中,n为Avrami 指数;Zt(10-4min-n)为结晶速率常数;Xt(%)为t时刻的相对结晶度;t(min)为结晶时间.对1-Xt= exp(-Zttn)进行对数处理,得到ln[-ln(1-Xt)]=lnZt+nlnt,其中,通过下式计算Xt:
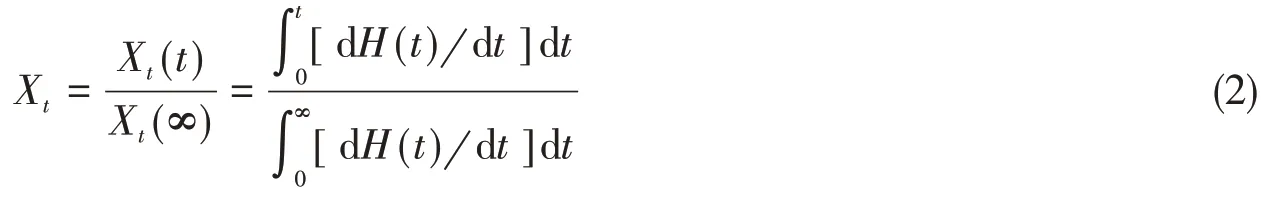
式中:dH(t)/dt(W/g·min)为结晶热流速率;Xt(t)(%)为t时刻的结晶度;Xt(∞)(%)为结晶过程完全结束后的结晶度;当Xt=0.5时,得到半结晶时间(t1/2,min):

2 结果与讨论
2.1 力学性能
由于GMA在接枝改性时易发生自聚等副反应,降低接枝产物的接枝率[26],因此,在PEBA接枝过程中,采用NVP作为共接枝单体来减少GMA接枝的副反应.图1(A)给出GMA和NVP分别作为接枝单体及共同添加对PEBA 进行改性时所得接枝产物(mPEBA)对PLA 复合材料冲击性能的影响.从图1(A)可以看出,当GMA和NVP分别作为接枝单体时,PLA/mPEBA(70/30,质量比)复合材料的冲击强度很低,与未接枝改性的PLA/PEBA(70/30,质量比)共混物的冲击强度相似,约为10 kJ/m2,说明单独使用两种单体对PEBA 进行接枝改性时无法对PLA 起到良好的增韧效果.当NVP 与GMA 协同对PEBA进行接枝改性时,PLA/mPEBA(70/30,质量比)共混物的冲击强度大幅度提高,高达73.8 kJ/m2.图1(B)示出了接枝单体组分用量对PLA/PEBA-GMA 共混体系冲击强度的影响.可见,当PEBA-GMA含量为10%时,接枝单体组分用量的变化对共混物的冲击强度影响不是很明显;当PEBA-GMA含量为20%时,共混体系的冲击强度随着接枝单体组分质量的增加而逐渐升高;当PEBA-GMA 含量为30%时,接枝单体组分用量的变化对冲击强度的影响非常显著,当接枝单体组分GMA,NVP,DCP 的用量分别2.5,2.5,0.25 g时,共混体系的冲击强度高达88.6 kJ/m2,具有超韧性能,表明接枝单体组分比例的提高能有效改善PEBA对PLA的增韧效果.
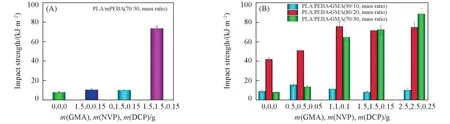
Fig.1 Impact strength of PLA/mPEBA and PLA/PEBA-GMA blends with different amounts of graft monomers
PLA/PEBA-GMA(70/30,质量比)共混体系的拉伸性能列于表1.接枝后PLA/PEBA-GMA(70/30,质量比)共混物的拉伸强度有所下降,且接枝单体组分用量的变化对拉伸强度的影响很小.但断裂伸长率随着接枝单体组分用量的增加有明显的提高,当接枝单体组分GMA,NVP 和DCP 的用量分别为2.5,2.5,0.25 g 时,断裂伸长率高达164.1%,是未接枝前的约4.5 倍.这主要是因为PEBA-GMA 上的环氧基团与PLA 的端基发生反应,使PEBA 与PLA 基体两相间的界面黏结性显著提升.因此,PLA复合材料的冲击强度和断裂伸长率显著提高,归因于PLA和PEBA-GMA之间反应相容性的增强.
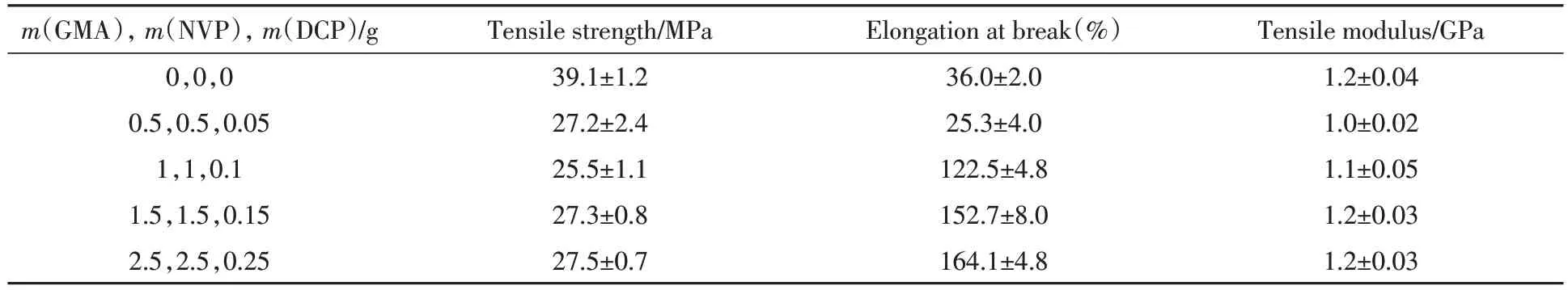
Table 1 Tensile properties of PLA/PEBA-GMA(70/30,mass ratio)blends with different amounts of graft monomers
2.2 结晶性能
为了考察PEBA-GMA 的接枝单体组分用量的变化对PLA 共混物结晶性能的影响,对PLA/PEBAGMA(70/30,质量比)共混物进行了非等温与等温结晶研究.从图2(A)的非等温结晶曲线可以看出,PLA/PEBA-GMA(70/30,质量比)共混物的冷结晶峰随着接枝单体组分用量的增加而逐渐减弱,表明PLA在一次降温中的结晶速率增加.对于PLA的双重熔融峰,接枝后高温熔融峰随着接枝单体组分用量的增加呈现了先减弱而后增强的趋势,PLA 的结晶度逐渐增加(表2),当接枝单体组分GMA,NVP和DCP 的用量分别为2.5,2.5,0.25 g 时,结晶度有明显提高,可达到11%.图2(B)为PLA/PEBAGMA(70/30,质量比)共混物在100oC 下的等温结晶曲线.可以看出,当接枝单体组分GMA,NVP 和DCP的用量分别为0.5,0.5,0.05 g时,PLA的半结晶时间(t1/2)高于纯PLA,而后随着接枝单体组分用量的增加,t1/2逐渐降低至3.9 min,表明接枝单体组分用量的增加能有效促进PLA的结晶.
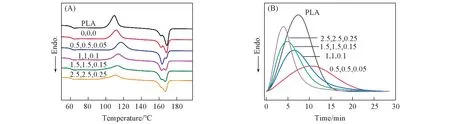
Fig.2 DSC curves of PLA/PEBA-GMA(70/30,mass ratio)blends with different amounts of graft monomer[m(GMA),m(NVP),m(DCP)/g]
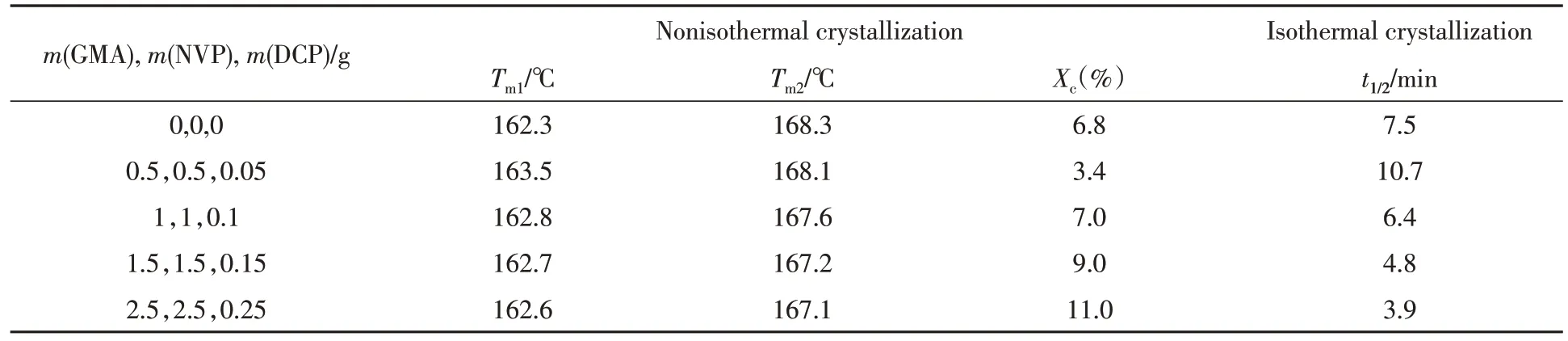
Table 2 DSC results of PLA/PEBA-GMA(70/30,mass ratio)blends with different amounts of graft monomers
2.3 界面形态
图3为PLA/PEBA-GMA(70/30,质量比)共混物液氮脆断面的SEM 照片.可以看出,未接枝的PEBA和未添加NVP接枝的PEBA-GMA在PLA基体中相区尺寸较大且分布不均匀,有剥离的孔洞和缝隙出现.当接枝单体组分GMA,NVP 和DCP 的用量分别为0.5,0.5,0.05 g 时,分散相的分散状况没有明显改善,说明此时共混体系的界面相容性仍然较差,黏结强度较低,这时冲击强度也比较低.当接枝单体组分GMA,NVP,DCP的用量大于1,1,0.1 g时,分散相粒子尺寸明显变小,分布更加均匀,部分断裂面上分散相边界模糊.共混体系分散相粒子尺寸、冲击强度与接枝单体组分GMA,NVP,DCP用量之间的关系列于表3.随着接枝单体组分用量的升高,分散相粒子尺寸逐渐降低,当接枝单体组分GMA,NVP,DCP 的用量从0.5,0.5,0.05 g升高到1,1,0.1 g时,分散相粒子的粒径由1.84 μm降到0.84 μm,此时共混物的冲击性能显著升高,由13.4 kJ/m2升高至64.7 kJ/m2,体系发生脆-韧转变.通常,两种聚合物之间的界面张力对于相形态有重要影响,并且界面相容性在降低界面张力、增加界面黏结性从而形成较好的分散形态方面起着主要作用.PEBA-GMA上的GMA基团可与PLA的端基(—OH,—COOH)发生反应,并且随着接枝单体组分用量的增加,反应程度增强,降低了PLA/PEBA-GMA(70/30,质量比)共混物的界面张力,在共混物组分之间的界面处形成新的接枝共聚物将增强相界面黏结性并抑制分散相聚集,因此,PLA共混体系冲击强度大幅度提高.
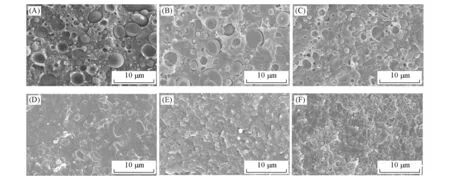
Fig.3 SEM image of freeze-fracture surface of the PLA/PEBA-GMA(70/30,mass ratio)blends with different amounts of graft monomers
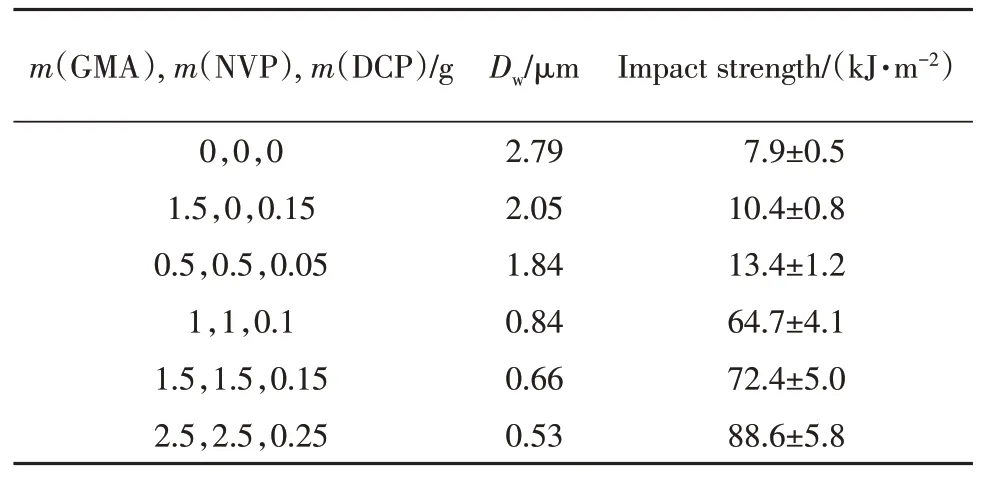
Table 3 Particle size of dispersed phase of PLA/PEBAGMA(70/30, mass ratio) blends with different amounts of graft monomers
2.4 流变性能
聚合物共混物的流变性能与其形态和组分之间的相互作用有关.图4示出了接枝单体组分用量的变化对PLA/PEBA-GMA(70/30,质量比)共混物复合黏度(η*)和储能模量(G′)的影响.低频区共混物的黏弹性响应可以反映聚合物组分之间的相互作用.因为在低频范围内,流动引起的分子取向对黏弹性几乎没有影响[27],但当两种聚合物熔融时,它们在界面处可能会有一些相互作用.当不加入接枝单体和加入接枝单体GMA,NVP 和DCP 的用量分别为0.5,0.5,0.05 g 时,在低频区,η*具有牛顿流体行为,而在高频区出现剪切变稀.随着接枝单体组分用量的增加,低频区共混体系的η*逐渐升高,这是由于接枝单体组分用量越高,PLA与PEBA-GMA的反应程度越高,在共混体系界面处形成新的接枝共聚物,使共混体系的分子量提高,导致η*升高.G′可用来表征复合黏度η*的弹性部分.从图4(B)可以看出,共混体系的G′均随着角频率(ω)的增加而升高.同时,随着接枝单体组分用量的增加,共混体系的储能模量在低频区逐渐上翘,这可能与分散相的相区尺寸降低以及在反应共混的过程中形成了准三维网络结构相关,说明分散相与基体的界面作用增强.
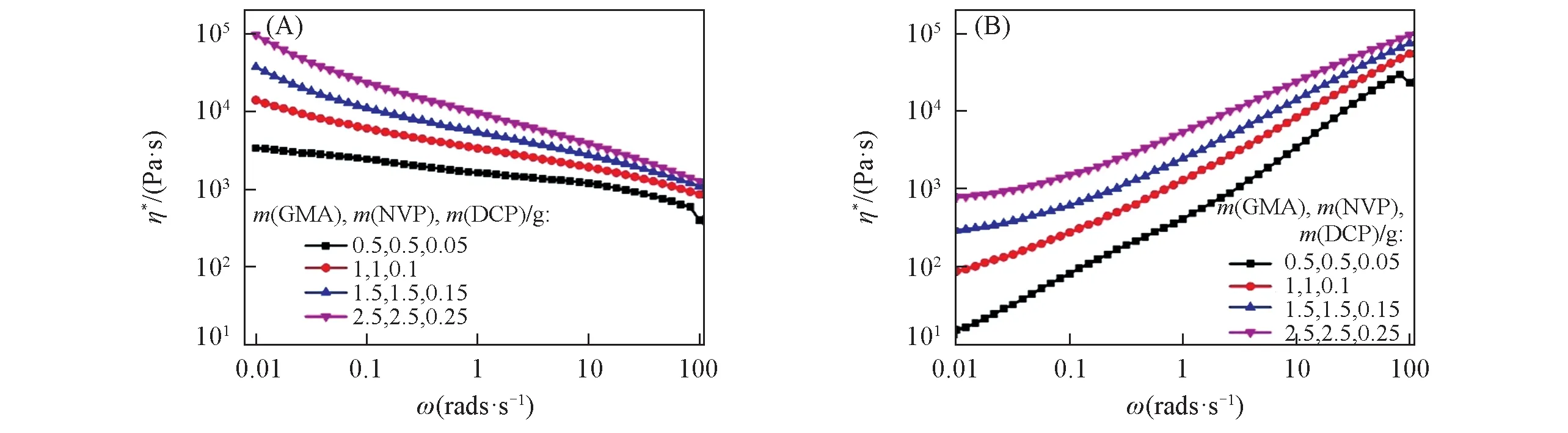
Fig.4 Rheology properties of PLA/PEBA-GMA(70/30,mass ratio)blends with different amounts of graft monomers at 180 ℃
2.5 增韧机理
为了证明PLA 与PEBA-GMA 之间的反应,将PLA/PEBA(70/30,质量比)和PLA/PEBA-GMA(70/30,质量比,其中PEBA-GMA中接枝单体组分GMA,NVP,DCP的用量分别为1.5,1.5,0.15 g和2.5,2.5,0.25 g)热压成50~100 μm的薄膜,然后在80oC的正丁醇中抽提24 h以除去未反应的PEBA(正丁醇是PEBA的良溶剂,PLA不溶于正丁醇中),抽提后的样品再次压成薄膜用于红外测试.图5为PLA,PEBA 和刻蚀后的PLA/PEBA(70/30,质量比)和PLA/PEBA-GMA(70/30,质量比)共混物的FTIR 谱图.可以看出,刻蚀后的PLA/PEBA(70/30,质量比)共混物的特征峰与纯PLA相似,没有出现PEBA的特征峰,表明在蚀刻过程中PEBA全部被去除掉,PEBA和PLA之间没有发生化学反应.而PLA/PEBA-GMA(70/30,质量比)共混物在3300,1539 和1640 cm-1处出现了PEBA 的特征峰,说明在刻蚀过程中PEBA组分没有被完全去除掉,表明在熔融共混过程中PLA端基(—OH,—COOH)与PEBA-GMA环氧基团之间发生反应并原位形成了新的接枝共聚物PLA-g-PEBA,使共混体系的界面相容性大幅度提高.当接枝单体组分GMA,NVP,DCP的用量分别为2.5,2.5,0.25 g 时,共混体系中PEBA 的特征峰与接枝单体组分质量分别为1.5,1.5,0.15 g 时相比显著增强,表明接枝单体组分用量的提高使PLA 与PEBA-GMA 的反应程度增强,界面黏结性更强,因此共混体系的冲击韧性更高.
PLA 与PEBA-GMA 在熔融反应共混过程中可能发生的反应如Scheme 1 所示.反应1 和反应2 表示PLA端基(—OH,—COOH)与PEBA-GMA上环氧基团之间的反应.在反应过程中,共混物的界面处形成新的接枝共聚物PLA-g-PEBA,其对有效改善共混体系界面相容性起着至关重要的作用,可以降低界面张力,改善分散相形态的均匀性,从而使分散相更好地分布在PLA基体中.同时,在反应共混过程中新形成的PLA-g-PEBA 还可以继续与PEBA-GMA 发生反应,如PLA-g-PEBA 共聚物的仲羟基可与PEBA-GMA的环氧基团进一步反应,生成的产物还可继续与PEBA-GMA发生反应形成交联网络结构.这与流变分析结果一致,PLA/PEBA复合材料在低频下具有较高的复合黏度.
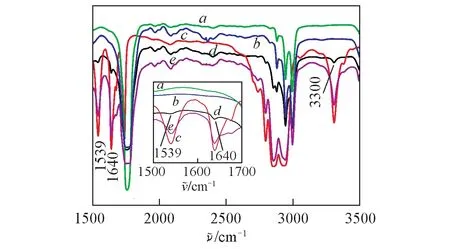
Fig.5 FTIR spectra of PLA(a), PLA/PEBA(b), PEBA(c), PLA/PEBA-GMA(1.5, 1.5, 0.15)(d) and PLA/PEBA-GMA(2.5,2.5,0.25)(e)
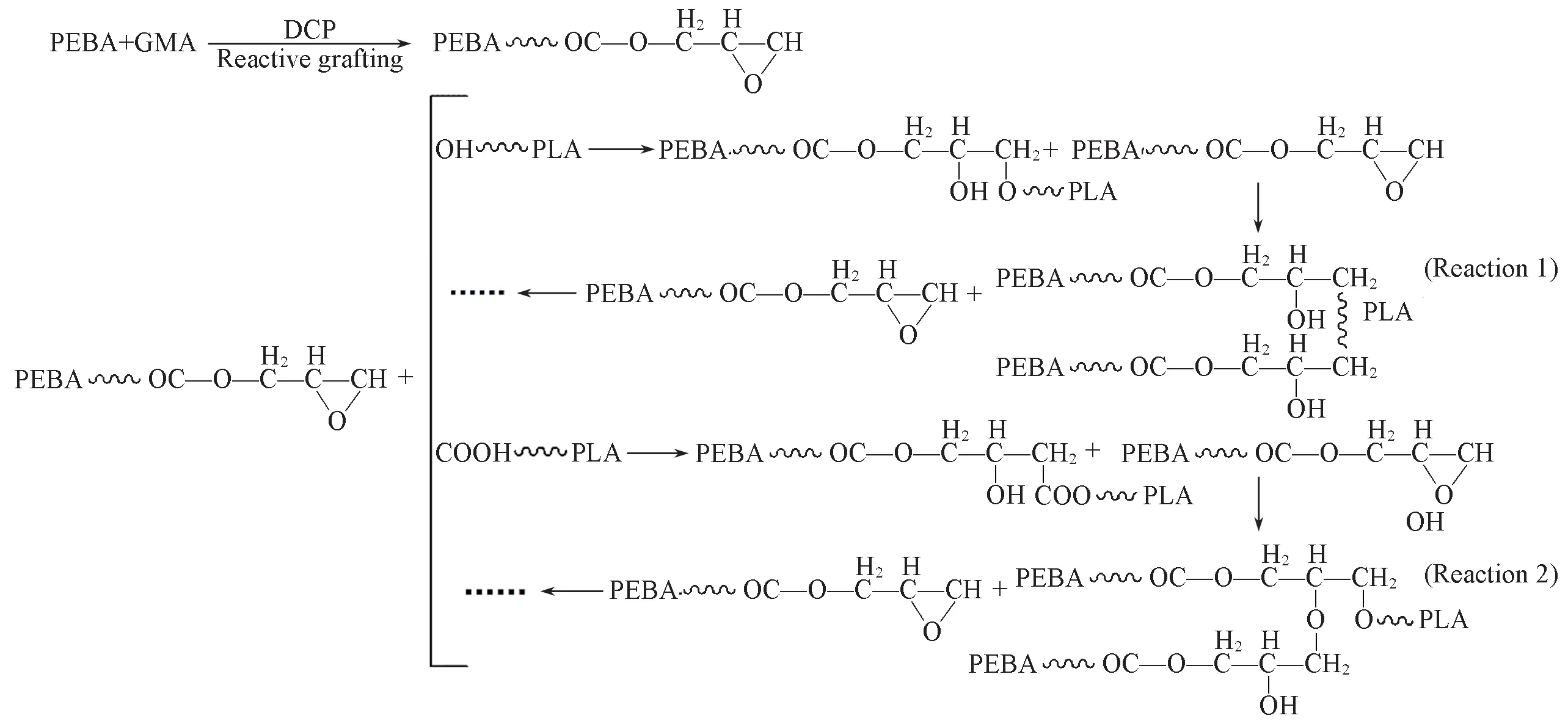
Scheme 1 Possible reactions between PLA and PEBA-GMA during reactive blends
综上所述,本文通过添加共聚单体NVP,使GMA 成功接枝得到PEBA 上,随着接枝单体组分GMA,NVP,DCP用量的增加,成功制备了冲击强度高达88.6 kJ/m2的超韧PLA/PEBA-GMA复合材料.接枝单体组分用量的增加可有效改善PLA与PEBA-GMA之间的界面相容性,使分散相在PLA基体中分布得更加均匀,因此,PLA/PEBA复合材料的力学性能大幅度提高.同时,PLA的结晶能力也得到有效改善.本文工作为制备全生物基超韧PLA复合材料提供了新思路.
猜你喜欢
学与玩(2022年12期)2023-01-11 06:39:22
合成树脂及塑料(2020年6期)2020-12-29 07:02:02
石油沥青(2019年4期)2019-09-02 01:41:54
中国塑料(2016年6期)2016-06-27 06:34:18
中国塑料(2016年3期)2016-06-15 20:30:03
中国塑料(2016年9期)2016-06-13 03:18:50
中国塑料(2015年7期)2015-10-14 01:02:40
中国塑料(2015年1期)2015-10-14 00:58:41
合成纤维工业(2015年4期)2015-03-25 12:52:38
火炸药学报(2014年1期)2014-03-20 13:17:24
