
南极利奇菲尔德岛土壤微生物多样性的初步分析
2020-08-26 01:57:48宋易洋李桂秀王国良
烟台大学学报(自然科学与工程版) 2020年3期
宋易洋,李桂秀,赵 芯,王国良
(1.烟台大学生命科学学院,山东 烟台 264005;2.北京市农林科学院北京农业生物技术研究中心,北京 100097)
南极是地球上的极端地区之一,其恶劣的生境条件孕育了特殊的土壤微生物类群[1-2].南极地区独特的地理位置和环境特征使大量土壤资源未被开发,其中包括许多具有特殊生理生化特性和分子生物学机制的微生物资源,例如大量具有耐/嗜盐、耐/嗜冷、抗辐射等特性的微生物[3].
自然界中的微生物只有很小一部分可在实验室进行分离培养,因此土壤菌株的分离实验结果无法描述土壤样品中微生物的多样性及物种之间的差异和相互作用关系[4].近年来,随着高通量测序技术的迅速发展,宏基因组测序成为可能[5].应用这种免培养的新技术,理论上可以获得样本中微生物的全部DNA信息,并且可以跳过微生物分离培养的过程,直接在基因水平揭示微生物的多样性与丰度,深入了解微生物的功能和相互作用,有助于进一步了解微生物群落的功能活动规律和遗传潜力[6-7].本研究以南极土壤为样本,采用宏基因组测序结果分析样本中微生物的多样性,为后续南极土壤微生物资源的开发和保护提供技术支持.
1 材料与方法
1.1 土壤样本采集
实验样本来自中国第31次南极科学考察(2014年10月30日出发),采样区域为利奇菲尔德岛,采样时间为2015年2月3日.所有样本低温条件运输至国内,实验前在-80 ℃条件下储藏于中国科学院北京基因组研究所.样本相关信息见表1.
1.2 实验方法
1.2.1 DNA提取 使用电子天平称取3例南极土壤样本各0.1 g,使用试剂盒(江苏康为世纪, cw2091s)对样本进行基因组提取.
1.2.2 DNA质量检测 宏基因组DNA的纯度和完整性检测分别通过NanoDrop微量分光光度计和琼脂糖凝胶水平电泳完成,DNA质量浓度通过Qubit检测获得.

表1 南极土壤样本位点信息
1.2.3 文库构建和高通量测序 采用二代测序技术进行高通量测序,需要对提取的宏基因组样本进行文库构建.取10 ng 基因组DNA起始建库,利用自动聚焦声波基因组剪切仪Covaris M220将gDNA打断成200 bp左右的片段.使用1.8×DNA 纯化磁珠(Vazyme, N411)纯化片段化产物,使用诺唯赞建库试剂(Vazyme, ND607)进行建库实验.分别用全自动核酸分析系统Qsep100和荧光定量PCR(BioRad, CFX96)对文库进行质检以确定其片段分布及质量浓度.质检合格的文库由北京贝瑞和康生物技术有限公司完成测序,测序基于Illumina NovaSeq 6000平台,测序策略为Paired-end 150 bp.
1.2.4 数据分析 测序得到的原始数据中,会带有接头序列或少量低质量的序列.使用FastQC对下机数据进行质控,生成质检报告.根据质检报告结果使用Trimmomatic[8]对数据进行过滤,以去除接头序列等低质量数据.然后使用MetaPhlAn2[9]进行后续分析,通过快速比对准确估计物种的相对丰度,得到微生物群落组成信息[10].使用GraPhlAn基于分类等级绘制可视化进化树,构建物种群落结构图[11].
2 结果与分析
2.1 宏基因组提取结果
由图1可见,3例宏基因组DNA样本主带均在10 kb以上且无明显杂带或降解.NanoDrop测得吸光度A260/A280(表2)均分布于1.8~2.0,说明纯度符合实验要求.基于Qubit Assay测得的DNA质量浓度(表2)进行后续文库构建.
2.2 文库构建结果
如图2所示,经Qsep100检测得到3例样本的文库片段分布.文库分布于300~500 bp范围内,峰值分别为404、411、409 bp,无引物二聚体污染,质量符合上机要求,将文库进行二代高通量测序.
2.3 测序与数据分析结果
下机数据详细信息见表3.使用MetaPhlAn2对3个样本的微生物群落组成进行分析,获得微生物多样性丰度汇总表(表4)和相对丰度热图(图3).由图3可知,样本S14、S16、S22均有明显的黑色区域,这些黑色区域所覆盖的细菌数量相对较大,且3个样本均具有不同的白色区域,从整体上看,样本间的差异性较大.群落间丰度分析显示:3个位点的土壤样本中既存在大量共有的土壤微生物类群,也存在一定比例的特有微生物类群.S14样本细菌丰度占91.31%,优势菌属是颗粒菌属(Granulicella),占比为40.81%.S16样本细菌丰度占89.56%,优势菌属是罗思河小杆菌属(Rhodanobacter),占比为24.37%.与S14和S22相比,S16样本中的特有菌属是亚硝化螺菌属(Nitrosospira). S22样本细菌丰度占59.84%,真菌丰度占40.16%,优势菌种是白色念珠菌(Candidaalbicans).与S14和S16相比,S22样本中的特有菌种是Rickettsiabellii.

表2 NanoDrop和Qubit检测基因组DNA结果
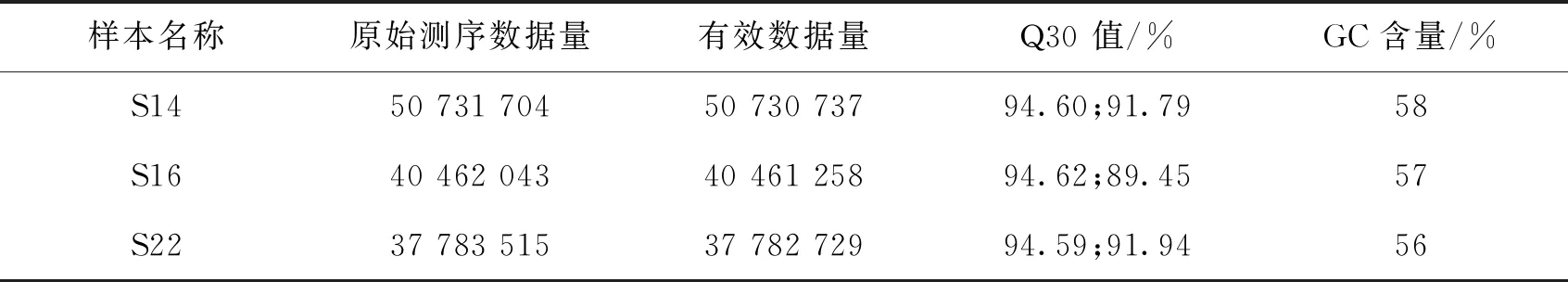
表3 测序数据统计结果
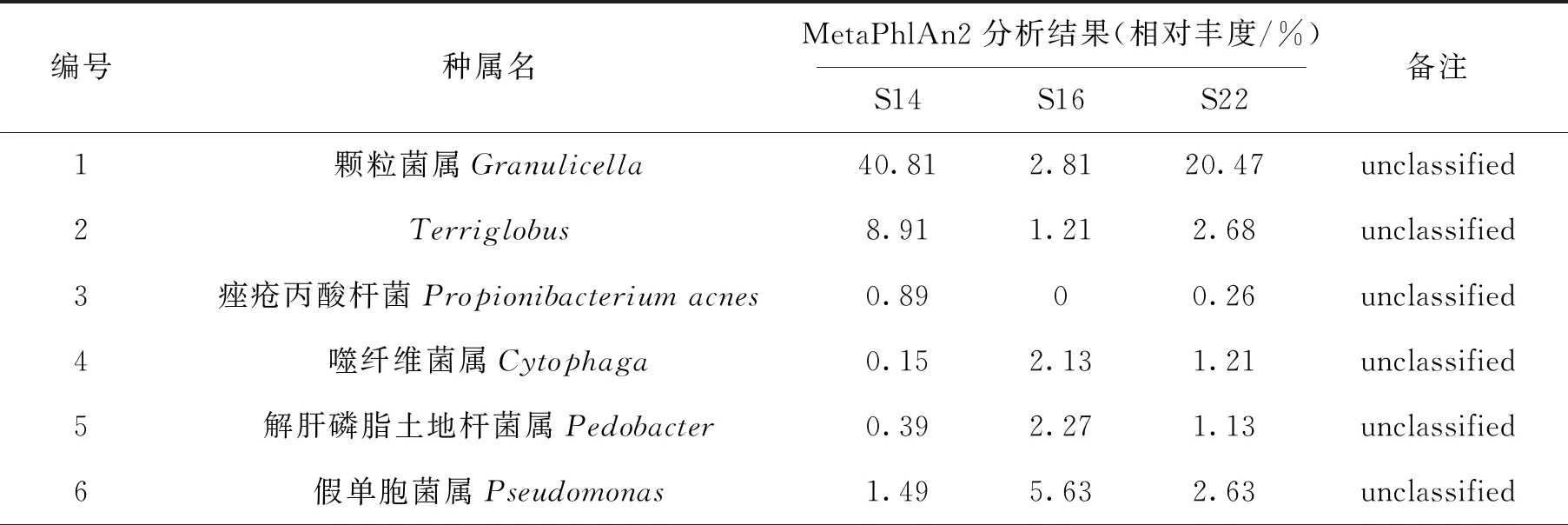
表4 微生物多样性分析结果
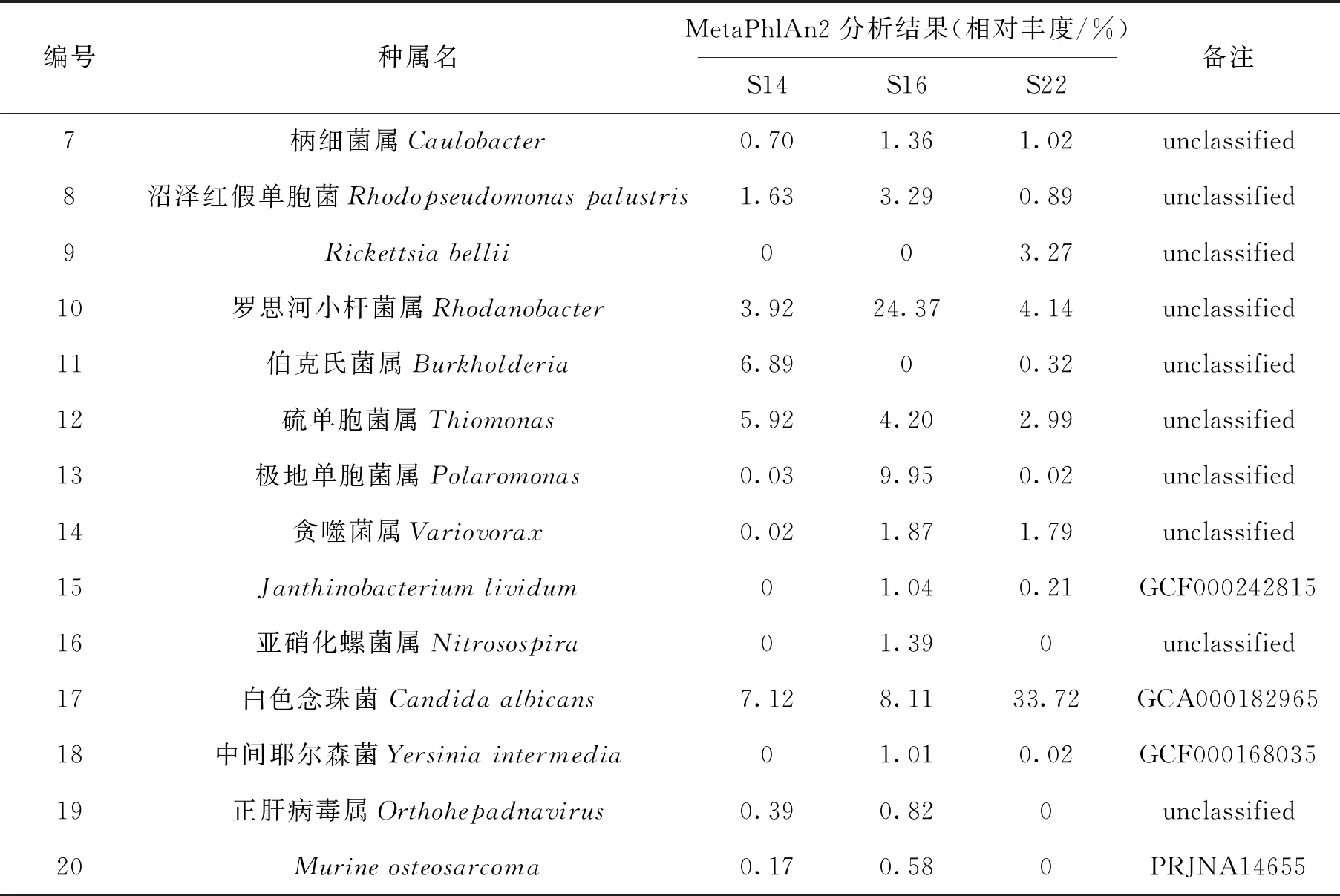
表4(续)
使用GraPhlAn生成物种群落结构图(图4),图4中从内而外的节点依次代表界门纲目科属种,节点越大,丰度越高.阴影区域表示不同目下的分类,同一目中的不同区域代表不同的科属种.图4中包含所有样本中相对丰度大于1%的15个目,从字母AB所属目为始,按逆时针顺序排列,分别为:酸杆菌目Acidobacteriales 、放线菌目Actinomycetales 、土壤红杆菌目Solirubrobacterales、噬纤维菌目 Cytophagales 、鞘脂杆菌目Sphingobacteriales、酵母目Saccharomycetales、Viruses noname、根瘤菌目Rhizobiales、柄杆菌目Caulobacterales 、立克次氏体目Rickettsiales、伯克氏菌目Burkholderiales、亚硝化单胞菌目Nitrosomonadales、肠杆菌目Enterobacteriales、假单胞菌目Pseudomonadales、黄单胞菌目Xanthomonadales.由图4可知,不同物种的丰度差异较大,酸杆菌目Acidobacteriales的物种丰度最高.
3 讨 论
采用高通量测序技术对南极3个不同位点的土壤微生物多样性进行分析,结果表明3个样本间的微生物种群丰度差异较大,优势菌属和特殊菌属各有不同.S14样本的优势菌属是颗粒菌属(Granulicella),Granulicella在赣南脐橙黄龙病植株和健康植株中都是优势菌属,并且是不可培养菌属[12].S16样本的优势菌属是罗思河小杆菌属(Rhodanobacter),在低pH的条件下,Rhodanobacter的反硝化细菌在地下污染严重的地区占主导地位,Rhodanobacter是耐酸的反硝化剂[13].S16样本中的特有菌属是亚硝化螺菌属(Nitrosospira),氨氧化细菌在硝化过程中起重要作用,Nitrosospira主导土壤中的氨氧化群落[14].S22样本的优势菌种是白色念珠菌(Candidaalbicans),Candidaalbicans是一株致病菌,它可以进入血液引起全身性疾病,几乎可以感染宿主的所有组织[15].S22样本中的特有菌种是Rickettsiabellii,Rickettsiabellii是一种专性细胞内细菌,是少数编码与细胞接合转移遗传元件相关的基因的立克次氏体之一[16-17].
MetaPhlAn2可以基于宏基因组数据,精确到微生物群体中种的构成,包括细菌、古菌、真核生物和病毒.若数据库中有株水平的基因组,也可以获得更加深入的研究.MetaPhlAn2整理了超过17 000个参考基因组,包括13 500个细菌和古菌,3 500个病毒和110个真核生物.MetaPhlAn2可以准确估计物种的相对丰度并鉴定到种,本研究中的部分微生物序列只鉴定到属,未精确到种,可能的原因是数据库中没有该微生物种水平的基因组数据,可以推测该微生物具有新种的可能性,侧面说明南极土壤中新种存在率高,具有较高的开发价值.研究表明,由于MetaPhlAn2具有更完善的病毒、真核生物和原核生物数据库,MetaPhlAn2比MetaPhlAn、mOTU[18]、Kraken[19]得到的分析结果更准确.
南极地区土壤样本由于位置特殊,运输至国内必须经过低温冷藏,很难实现对新鲜样本进行微生物群落分析.本实验通过宏基因组高通量测序技术对冷冻后的土壤样本中的微生物群落构成信息进行鉴定,为南极土壤微生物资源开发提供了信息储备和技术支持.
当前对微生物互作的代谢调控通路分析和构建多数是基于单一种群,但自然界中微生物群落是以复杂多样的整体形式存在的.因此将微生物种群进行整体代谢调控网络和通路的分析研究甚至加以改造和利用将是一个新的发展方向.高速发展的宏基因组高通量测序技术和分析手段为有效探索微生物群落的种群构成和互相作用的调控网络提供了强大的技术支持.
猜你喜欢
国际太空(2023年1期)2023-02-27 09:03:42
猪业科学(2021年3期)2021-05-21 02:05:36
小哥白尼(趣味科学)(2020年12期)2021-01-18 06:15:24
小哥白尼(神奇星球)(2020年12期)2021-01-18 05:36:06
透析与人工器官(2020年1期)2020-11-16 01:42:34
幽默大师(2020年10期)2020-11-10 09:07:22
中华诗词(2019年1期)2019-11-14 23:33:56
铁道通信信号(2019年8期)2019-10-10 05:06:00
学生天地(2019年32期)2019-08-25 08:55:20
猪业科学(2018年4期)2018-05-19 02:04:31
