
2-羟甲基氯雷他定和4-羟甲基氯雷他定的合成路线研究
2020-08-25 01:52徐理张泽宇贺美
当代化工 2020年5期
徐理 张泽宇 贺美

摘 要: 氯雷他定糖浆制剂生产和存储过程中会产生的两种杂质(2-羟甲基氯雷他定Ⅱ和4-羟甲基氯雷他定Ⅲ)。两种杂质的成功合成将有助于氯雷他定的杂质去除以及用药安全的相关研究。以氯雷他定(Ⅰ)为原料氧化生成N-氧化物Ⅳ,然后转化为N-甲氧基吡啶鎓盐Ⅴ,再经亲核反应生成腈化物Ⅵ和Ⅶ的混合物,Ⅵ和Ⅶ是生成物中的主要成分(4∶1)。Ⅵ和Ⅶ通过Pinner反应生成酯Ⅷ和Ⅸ,经DIBAL-H还原后得到目标产物2-羟甲基氯雷他定Ⅱ与4-羟甲基氯雷他定Ⅲ。2-羟甲基氯雷他定和4-羟甲基氯雷他定的结构经1H-NMR谱和MS谱证实,纯度经HPLC测定分别为99.38%和99.41%。
关 键 词:氯雷他定;2-羟甲基氯雷他定;4-羟甲基氯雷他定;抗组胺;杂质
中图分类号:TQ031.2 文献标识码: A 文章编号: 1671-0460(2020)05-0766-04
Abstract: The formation of two impurities of loratadine (2-hydroxymethyl loratadine Ⅱ and 4-hydroxymethyl loratadine Ⅲ) generated in producing process of loratadine syrup, was fully revealed. The successful synthesis of 2- hydroxymethyl loratadine and 4-hydroxymethyl loratadine can facilitate the impurity removal and medical-safety studies of loratadine. The crucial steps of this synthesis route were converting loratadineⅠinto the N-oxide Ⅵ, and then into the N-methoxypyridinium salt Ⅴ. The attack of cyanide ion produced the mixture of the corresponding nitriles Ⅵ and Ⅶ, with the 2-isomer predominated of 4∶1. Then Ⅵ and Ⅶ were converted into ester Ⅷ and Ⅸ by classical Pinner process, which was reduced by DIBAL-H to procudce Ⅱ and Ⅲ, respectively. 2-Hydroxymethyl loratadine and 4-hydroxymethyl loratadine were obtained, the chemical purity was determined as 99.38% and 99.41% by HPLC,and their structures were confirmed by 1H-NMR and MS, respectively.
Key words: Loratadine; 2- and 4-hydroxymethyl loratadine; Antihiatamine; Impurities
氯雷他定(loratadine,Ⅰ),化學名称 “4-(8-氯-5,6-二氢-11H苯并[5,6]环庚并[1,2-b]吡啶-11-亚基)-1-哌啶羧酸乙酯”,由美国Schering-Plough公司自主研发,于1988年3月在比利时首先上市[1,2]。氯雷他定是一种非镇静性的抗组胺药,具有治疗过敏性鼻炎、急性或慢性荨麻疹、过敏性结膜炎、花粉症及其他过敏性疾病的作用,且氯雷他定具有起效快、作用强、持续时间长、无中枢神经镇静作用等特点,是人们治疗过敏性疾病的首选用药[3]。
因氯雷他定需要以糖浆制剂的形式供儿科患者服用,故市面上保有相当数量的氯雷他定糖浆制剂(内含柠檬酸、甘油、丙二醇、苯甲酸钠、蔗糖等,pH为2.8)。而研究显示,在生产和存储糖浆制剂的过程中,均会检测出几种氯雷他定的衍生物,主要是2-羟甲基氯雷他定(2-Hydroxymethyl loratadine,Ⅱ)和4-羟甲基氯雷他定(4-Hydroxymethyl loratadine,Ⅲ)[4]。Eyjolfsson推测了氯雷他定在糖浆制剂中发生羟甲基化生成Ⅱ 和Ⅲ的化学路线(见图1),即Ⅰ被氧气氧化,生成了N-氧化物Ⅳ。同时,制剂中的蔗糖水解成葡萄糖与果糖,后者可在酸催化脱水过程中通过逆Aldol反应生成2-呋喃甲醛和甲醛。此外,果糖可能形成5-(羟甲基)-2-呋喃甲醛,其作为甲醛供体也发挥了作用。然后,上述甲醛等亲电试剂会攻击Ⅳ中吡啶环上的富电子的2-位和4-位,生成Ⅹ和Ⅺ。最后Ⅹ和Ⅺ经还原生成Ⅱ和Ⅲ[5]。但是,上述路线的Ⅱ、Ⅲ产率极低,不适合作为合成Ⅱ、Ⅲ的实际应用路线。
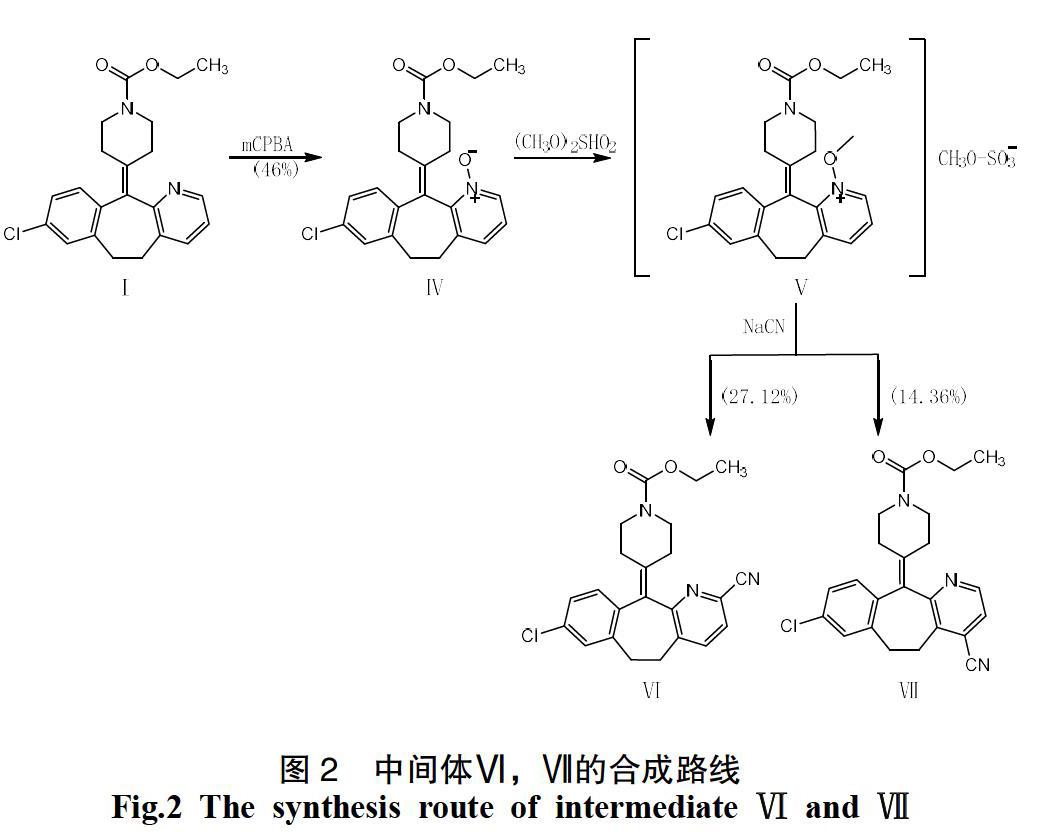
因此,需要设计出易操作、收率更高的反应路线。本实验参考Cerrada[6]、Okamota和Tani[7]、Feely和Beavers-Tani[8]的策略设计合成路线,将起始物氯雷他定Ⅰ氧化生成N-氧化物Ⅳ,然后加入硫酸二甲酯转化为N-甲氧基吡啶鎓盐Ⅴ。
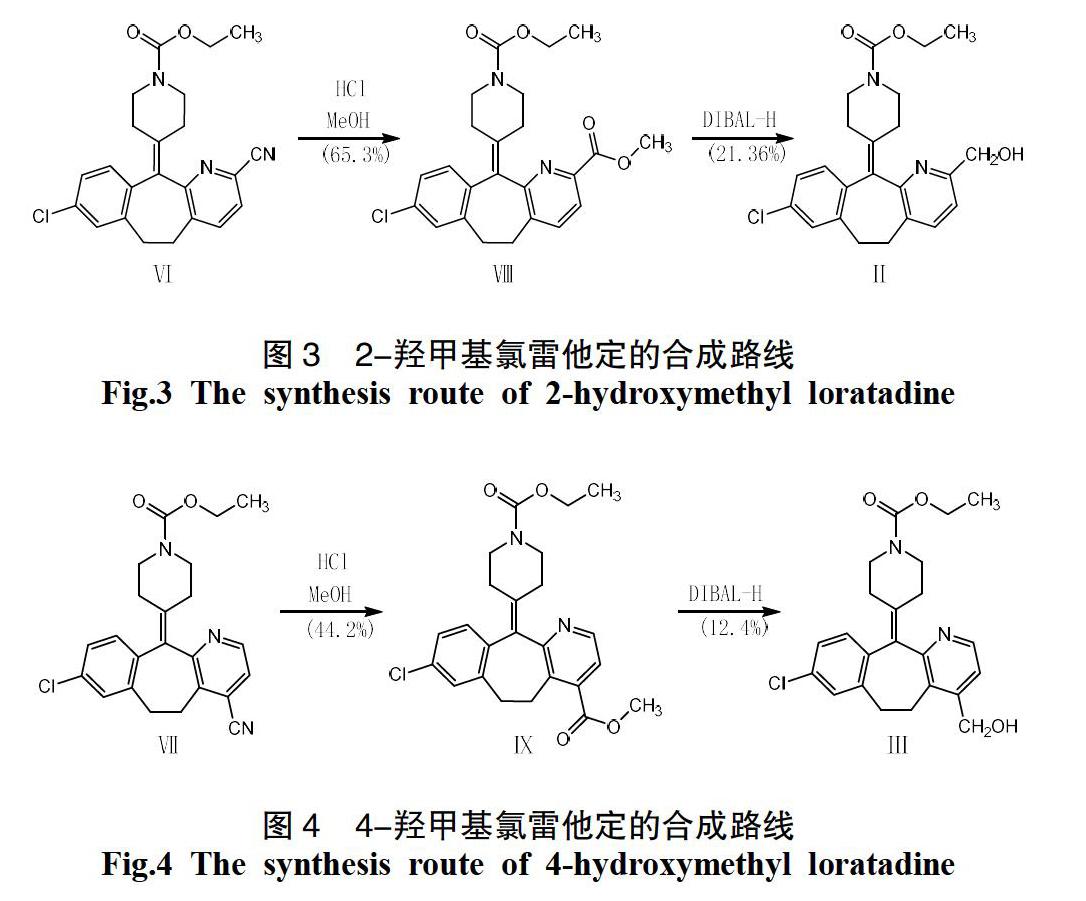
再用氰根离子进攻Ⅴ生成Ⅵ和Ⅶ的混合物,两种产物均通过色谱法分离,并用于制备最终化合物(见图2)。之后,通过对Ⅵ进行Pinner反应生成Ⅷ,再通过二异丁基氢化铝还原后合成目标产物Ⅱ(见图3);同理可通过Ⅶ生成Ⅸ,最后合成目标产物Ⅲ(见图4)。
1 仪器试剂
熔点采用XT-4型显微熔点测定仪测定,温度未经校正。1H-NMR谱采用Bruker ARX-300型核磁共振波谱仪测定,TMS为内标。质谱采用Water Quanttro micro API三重四级杆串联质谱仪测定。化合物纯度采用Agilent 1200高效液相色谱仪测定[色谱柱XDB-C18柱(4.6 mm×150mm,5μm)]。所用溶劑均为分析纯,使用时根据需要进一步干燥。
2 实验过程
2.1 4-(8-氯-1-氧基-5,6-二氢苯并[5,6]环庚[1,2-b]吡啶基-11亚烷基)哌啶-1-甲酸乙酯(Ⅳ)的合成
将氯雷他定1.125 g(0.064 35 mol)溶解在CH2Cl2(60 mL)中。一次性加入mCPBA(4.14 g, 0.019 5 mol),在60 ℃下搅拌25 h。TLC(展开剂:EA∶PE∶MeOH =1∶1∶0.1)显示反应完全。冷却至室温,蒸干溶剂,得到黏稠的粗品。粗品经硅胶柱层析(VEA∶VPE∶VMeOH=100∶10∶1),得到白色固体Ⅳ(3.6 g,46.0%)。mp 139~141 ℃,1H-NMR(CDCl3)σ1.25(t,3H,CH3),2.25~2.38(m,2H,CH2),2.46~2.59(m,2H,CH2),2.75~2.96(m,2H,CH2), 3.3.~3.47(m,4H,2×CH2),3.64~3.84(m,2H,CH2),4.11(m,2H,CH2),7.06~7.14(m,4H,Ar-H),7.20~7.26(m,1H,Ar-H),8.08(t,1H,Ar-H)。
2.2 4-(8-氯-2-氰基-5,6-二氢苯并[5,6]环庚[1,2-b]吡啶-11-亚基)哌啶-1-甲酸乙酯(Ⅵ)和 4-(8-氯-4-氰基-5,6-二氢苯并-[5,6]环庚[1,2-b]吡啶-11-亚基)哌啶-1-甲酸乙酯(Ⅶ)的合成
将化合物Ⅳ(91.5 g,0.229 35 mol)溶于丙酮(2 750 mL)中,恒温60 ℃搅拌,滴加硫酸二甲酯(28.935 mL,0.229 35 mol)。滴毕,60 ℃搅拌一夜。TLC1(展开剂:EA∶PE=1∶1)显示反应完全。蒸干溶剂,得到棕色油状吡啶鎓盐Ⅴ。无须进一步纯化即可用于下一步。
将NaCN(22.5 g,0.459 mol)溶解在水(300mL)中,并缓慢加入溶解在水(900 mL)中的Ⅴ(29.79 g,0.225 mol)。室温搅拌10 min,TLC2(展开剂:EA∶PE=1∶3)显示反应完全。用乙醚萃取,有机层经Na2SO4干燥,过滤,浓缩至干,并将粗品经硅胶柱层析(VEA∶VPE∶VMeOH=100∶10∶1),得到白色固体Ⅵ(25.5 g,27.12%)和白色固体Ⅶ(13.5 g,14.36%)。
6:mp 193~195 ℃,1H-NMR(CDCl3)σ1.26(t,3H,CH3),2.19~2.55(m,4H,2×CH2),2.77~2.98(m,2H,CH2),3.16~3.29(m,2H,CH2),3.36~3.47(m,2H,CH2),3.72~3.78(m,2H,CH2), 4.15(m,2H,CH2),7.09~7.20(m,3H,3×Ar-H),7.47~7.59(m,2H,2×Ar-H);7:mp 99~100 ℃,1H-NMR(CDCl3)σ1.25(t,3H,CH3),2.37~2.34(m,4H,2×CH2),3.34~3.82(m,4H,2×CH2),3.38~3.51(m,2H,CH2),3.72~3.90(m,2H,CH2),4.17(m,2H,CH2),7.12~7.16(m,3H,3×Ar-H),7.39(d,1H,Ar-H),8.57(d,1H,Ar-H)。
2.3 8-氯-11-(1-乙氧基羰基哌啶-4-亚烷基)-6,11-二氢-5H-苯并[5,6]环庚-[1,2-b]吡啶-2-羧酸甲酯(Ⅷ)的合成
将化合物Ⅵ(25.5 g,0.062 55 mol)溶于CH2Cl2(540 mL)和甲醇(540 mL)的混合溶液中,冷却至0 ℃,鼓入HCl气体5 h。TLC1(展开剂EA/PE=1:2)显示反应完全。室温搅拌15小时。TLC2(展开剂EA/PE=1:3)显示反应完全。将溶剂蒸发至干,将残余物用水(150mL)洗涤,用CH2Cl2(3×75 mL)萃取。分离有机层,经Na2SO4干燥,过滤并浓缩至干。残余物通过乙醇重结晶,得到白色固体Ⅷ(18 g,65.3%)。mp 145~148 ℃. 1H-NMR(CDCl3)σ1.25(t,3H,CH3),2.28~2.53(m,4H,2×CH2),2.77~2.96(m,2H,CH2),3.08~ 3.15(m,2H,CH2),3.35~3.44(m,2H,CH2), 3.75~3.88(m,2H,CH2),3.95(s,3H,CH3), 4.13(m,2H,CH2),7.12~7.21(m,3H,3×Ar-H),7.57(d,1H,Ar-H),7.91(d,1H,Ar-H)。
2.4 4-(8-氯-2-羟甲基-5,6-二氢苯并[5,6]环庚[1,2-b]吡啶-11-亚烷基)-哌啶-1-甲酸乙酯(Ⅱ)的合成
将化合物Ⅷ(11.25 g,0.025 5 mol)溶于CH2Cl2(375 mL)中,氮气冷却至-10 ℃。滴加DIBAL-H(105 mL,0.105 mol)。滴毕,-10 ℃搅拌3 h,TLC(展开剂:EA∶PE=1∶3)显示反应完全。
将混合物倒入饱和的NH4Cl溶液(150 mL)中,通过CH2Cl2(2×150 mL)萃取。分离有机层,经Na2SO4干燥,过滤并浓缩至干。粗品经硅胶柱层析(V EA∶V PE=2∶1),得到白色固体Ⅱ(2.25 g, 21.36%)。mp 88~90 ℃,1H-NMR(CDCl3) σ 1.23~1.43(m,3H,CH3),2.28~2.38(m,2H,CH2),2.45~2.50(m,2H,CH2),2.77~2.91(m,2H,CH2),3.16~3.25(m,2H,CH2),3.31~3.41(m,2H,CH2), 3.70~3.79(m,2H,CH2),4.15(m,2H,CH2), 4.70(s,2H,OH),7.04~7.19(m,4H,4×Ar-H),7.44(d,1H,Ar-H). EHMS m/z:413.1([M+1]+,100). HPLC法(XDB-C18色谱柱;流动相:MeOH∶CH3COONH4=75∶25;流速:1.0 mL·min-1,检测保留时间为4.364 min,纯度为99.38%。
2.5 8-氯-11-(1-乙氧基羰基哌啶-4-亚烷基)-6,11-二氢-5H-苯并[5,6]环庚[1,2-b]-吡啶-4-羧酸甲酯(Ⅸ)的合成
将化合物Ⅶ(13.5 g,0.0331 5 mol)溶解于CH2Cl2(285 mL)和甲醇(285 mL)的混合溶液中。冷却至0 ℃,鼓入HCl气体5 h,TLC1(展開剂:EA∶PE=1∶2)显示反应完全。室温搅拌15 h。TLC2(展开剂:EA∶PE=1∶3)显示反应完全。
将溶剂蒸发至干,将残余物用水(75 mL)洗涤,用CH2Cl2(3×75 mL)萃取。分离有机层,经Na2SO4干燥,过滤并浓缩。残余物通过乙醇重结晶得到白色固体Ⅸ(6.45 g,44.2%)。mp 150~153 ℃,1H-NMR(CDCl3)σ1.24(t,3H,CH3),2.34~2.45(m,4H,2×CH2),2.97~3.03(m,2H,CH2),3.11~3.21(m,2H,CH2),3.34~3.47(m,2H,CH2),3.50~3.78(m,2H,CH2),3.93(s,3H,CH3),4.13(m,2H,CH2),7.09(m,3H,3×Ar-H),7.48(d,1H,Ar-H),8.48(d,1H,Ar-H)。
2.6 4-(8-氯-4-羟甲基-5,6-二氢苯并[5,6]环庚[1,2-b]吡啶基-11亚基)-哌啶-1-甲酸乙酯(Ⅲ)的合成
将化合物Ⅸ(6.45 g,0.014 7 mol)溶于CH2Cl2(195 mL)中,氮气冷却至-10 ℃,滴加DIBAL-H(60 mL,0.06 mol)。滴毕,-10 ℃搅拌混合物3 h。TLC(展开剂:EA∶PE=1∶3)显示反应完全。
将混合物倒入饱和的NH4Cl溶液(150 mL)中。通过CH2Cl2(2×150 mL)萃取。分离有机层,经Na2SO4干燥,过滤并浓缩至干。粗品经硅胶柱层析(V EA∶V PE=2∶1),得到的白色固体Ⅲ(0.72 g,12.4%)。mp 83~85 ℃,1H-NMR(CDCl3)σ1.31(t,3H,CH3),2.28~2.5(m,4H,2×CH2),2.78~2.90(m,2H,CH2),3.02~3.28(m,2H,CH2),3.31~3.49(m,2H,CH2),3.70~3.91(m,2H,CH2), 4.15(m,2H,CH2),4.70(s,2H,OH),7.13(m,3H,3×Ar-H),7.22~7.28(m,1H,Ar-H),8.43(d,1H,Ar-H); EHMS m/z:413.1([M+1]+,100)。HPLC法(XDB-C18色谱柱;流动相:MeOH∶CH3COONH4=75∶25;流速:1.0 mL·min-1,检测保留时间为4.364 min,纯度为99.41%。
3 结 论
综合国内外的相关研究,提出了一条较为适用的2-羟甲基氯雷他定(Ⅱ)和4-羟甲基氯雷他定(Ⅲ)的合成方法,通过纯化使这两个杂质的质量分数均大于99%。利用1H-NMR确认了结构,通过HPLC确定了纯度[9,10]。本研究对建立并完善氯雷他定的分析方法、制定已知杂质的限度、提高药品品质标准,都具有重要的意义。
由中间产物Ⅷ和Ⅸ制备目标产物Ⅱ、Ⅲ的过程,文献[6]采用了将DIBAL-H分三次滴加,每次滴毕后回流加热16 h的方法,最后目标产物Ⅱ、Ⅲ的收率均为12%;本工艺路线采用一次性加入DIBAL-H后在-10℃下搅拌3 h,加入饱和的NH4Cl溶液进行淬灭,再进行萃取的方法。目标产物Ⅱ、Ⅲ的收率分别为21.36%和12.4%。在这一步中,本工艺路线大幅节省了时间并提高了收率,对终产物Ⅱ、Ⅲ的合成以及相关研究的开展有着十足的参考意义。
参考文献:
[1]Villani,F. J. Antihistaminic11-(4-piperidylidene)-5H-benzo-[5,6]-cyclohepta-[1,2-b]-pypridines:US,4282233[P]. 1981.
[2]Radwanski,E.,Hilbert,J.,Symchowicz,S.,et al. Loratadine: multiple-dose pharmacokinetics[J].Journal of clinical pharmacology ,2013,27(7): 530-533.
[3]贾鹏飞,王娟,李晓林,韩晓敏. 超声波辐射技术合成氯雷他定[J]. 精细化工,2017(10): 99-103.
[4]Munayyer,F. J., Guazzo,F., Stupak,E. I., et al. Stablilized antihistamine syrup: US, 6132758[P].2000.
[5]Eyjolfsson,R. Loratadine: hydroxymethylation in syrup[J]. Pharmazie,2003,5: 154-154.
[6]Cerrada,V.,Matía-Martín,M. P.,Novella,J. L.,et al. Synthesis of 2-and 4-hydroxymethyl Loratadine, usual impurities inLoratadine syrup formulations[J].Arkivoc Archive for Organic Chemistry,2005(9): 200-206.
[7]Okamoto,T.; Tani,H. Reaction mechanism in aromatic heterocyclic compound. I. The reactions of N-alkoxypyridinium derivarives[J].Chem. Pharm. Bull,1959,7:130-130.
[8]Feely,W. E.; Beavers,E. M. Cyanation of amino oxide salts. A new synthesis of cyanopyridines[J].J. Am. Chem. Soc.1959,81:4004-4007.
[9]唐学红,肖先举. 高效液相色谱-质谱联用技术在药物分析中的应用[J]. 当代化工,2011,40(9): 988-990.
[10]吕孝丽,徐烨,雷雅娟. 高效液相色谱法同时测定饮料中的着色剂、甜味剂与防腐剂[J]. 当代化工,2007(2): 98-101.
猜你喜欢
学苑创造·B版(2019年11期)2019-12-05
佛山陶瓷(2018年6期)2018-09-14
小猕猴智力画刊(2018年3期)2018-06-12
小猕猴智力画刊(2018年4期)2018-06-09
中外医疗(2017年35期)2018-03-07
理科考试研究·高中(2017年7期)2017-11-04
中国医药导报(2017年2期)2017-03-18
小学生导刊(低年级)(2016年8期)2016-09-24
江苏农业科学(2014年10期)2014-11-22
中学理科·综合版(2008年11期)2008-01-14
