
黑龙江省表层冻土细菌群落结构组成和功能特征
2020-08-18 07:24关健飞
生态学报 2020年14期
关健飞,曹 阳
牡丹江师范学院历史与文化学院, 牡丹江 157000
受全球气候变化影响,中国平均气温变化趋势与全球变化趋势一致,而东北地区气温升温幅度高于全国的平均升温幅度[1],是增温最快的地区之一[2]。东北多年冻土区是欧亚大陆多年冻土区的南缘地带,冻土赋存条件脆弱,热稳定性差,易受气候和外界环境变化的影响,生态系统敏感性强[3]。位于东部季风区北部的黑龙江省纬度位置偏北,受强大的西伯利亚、蒙古高压影响形成严寒的大陆性气候,形成和发育创造了不同类型的冻土和冻土特征。黑龙江省多年冻土分布于47°30′—53°33′N,分为三个亚区,分别为大兴安岭北部大片多年冻土亚区、大兴安岭中部大片岛状多年冻土亚区以及大、小兴安岭岛状-稀疏岛状多年冻土亚区;其余地区为季节性冻土区[4]。
冻土,一般是指温度在0℃或0℃以下,并含有冰的各种岩土和土壤。冻土作为一种温度敏感和易变的地质体,是气候变化的敏感区,气候变化是影响冻土的重要因素,微生物对于预测冻土和气候之间的潜在反馈机制至关重要[5-6]。微生物作为冻土生态系统中的重要角色扮演者,是其内部多种功能联通的生态群体[7]。冻土微生物在冻土生物地球化学循环中(如土壤碳氮循环、温室气体排放等)具有重要的作用[8- 10],且在一定程度上能够指示全球气候变化。在全球气候变暖的背景下,研究冻土微生物群落特征及其与环境因子变化的关系逐渐成为当前热点研究领域[11]。微生物的种群结构和遗传进化对环境条件变化敏感,研究不同冻土区土壤中微生物的多样性和种群结构将有助于及时检测环境变化并采取有效的应对措施[12]。环境因子被认为是影响微生物群落发生变化的主要驱动力[13],如土壤pH[14]、气候条件[15]、有机质含量[16]等都可以在一定程度上影响微生物群落的组成和分布。
土壤微生物数量巨大、结构复杂,传统的微生物培养计数仅仅能够分离培养1%的微生物,很难全面、准确的解析土壤微生物的结构组成和多样性变化规律[17-19]。随着分子生物学技术的不断发展,高通量测试分析技术作为微生物群落的有效研究手段,被广泛的应用在土壤微生物研究领域,其中包括物种的多样性、丰富度、种内/种间相互作用、对环境变化响应等方面的研究[20-21]。本研究以黑龙江省表层冻土样品为研究对象,通过高通量分子测序手段,分析黑龙江省表层冻土细菌群落组成和分子生态网络特征,预测群落功能,探究环境因素、利用方式对于黑龙江表层冻土微生物群落空间异质性的影响。
1 材料与方法
1.1 研究区概况
黑龙江省,介于北纬43°26′—53°33′,东经121°11′—135°05′,是气候变化导致气温升高幅度最明显的地区之一。全省年平均气温多在- 5℃—5℃之间,由南向北降低,大致以嫩江、伊春一线为0℃等值线。无霜冻期全省平均介于100—150 d之间,终霜冻在4月下旬至5月上旬结束。年降水量全省多介于400—650 mm之间,中部山区多,东部次之,西、北部少。其北部的多年冻土属欧亚大陆多年冻土区的南缘地带,冻土类型主要为多年冻土和季节性冻土。
1.2 采样点布设及样品采集
本研究以黑龙江省表层(0—20 cm)冻土为研究对象,全省区域内共设置采样点19个(如图1所示),于2019年5月1—4日期间完成土壤样品采集。各采样点处设置10 m×10 m样地,四角及对角线交点处分别进行表层土壤样品的采集,去除杂物、细跟等杂物后充分混匀,置于密封袋中,带回实验室后,一部分- 80℃保存用于高通量测试分析,一部分常温风干用于土壤理化性质测定。

图1 采样点分布
1.3 研究方法
1.3.1土壤微生物群落测定
采用E.Z.N.A.® soil DNA Kit(Omega Bio-tek,Norcross,GA,U.S.)提取土壤总DNA,应用NanoDrop2000进行DNA纯度和浓度检测,DNA完整性检测采用琼脂糖凝胶电泳法(1%琼脂糖胶,5V/cm,20min)。采用16S rRNA基因的V3-V4区引物515F(5′-GTGCCAGCMGCCGCGG- 3′)和907R(5′-CCGTCAATTCMTTTRAGTTT- 3′)对高变区片段进行扩增。PCR反应条件为95℃预变性3min;27次循环(95℃变性30 s,55℃退火30 s,72℃延伸45 s);72℃延伸10 min,至10℃。20 μL反应体系为4 μL的5×FastPfu Buffer,2 μL的2.5 mM dNTPs,0.8 μL的Forward Primer(5 μM),0.8 μL的Reverse Primer(5 μM),0.4 μL的FastPfu Polymerase,0.2 μL的BSA,10 ng的Template DNA,补ddH2O至20 μL。使用2%琼脂糖凝胶回收PCR产物,利用AxyPrep DNA Gel Extraction Kit(Axygen Biosciences,Union City,CA,USA)进行纯化,Tris-HCl洗脱,2%琼脂糖电泳检测。利用QuantiFluorTM-ST进行检测定量。根据Illumina MiSeq平台标准操作规程将纯化后的扩增片段构建文库,利用Illumina公司的Miseq PE300平台进行测序,委托上海美吉生物医药科技有限公司完成。
1.3.2土壤理化性质测定
土壤含水率的测定采用真空烘箱法(NY 525- 2012/5.6);pH的测定采用电位法(NY/T 1377- 2007);有机质的测定采用重铬酸钾滴定法(NY/T 1121.6- 2006);全氮的测定采用半微量开氏法(NY/T 53- 1987);全磷的测定采用氢氧化钠碱融-钼锑抗比色法(NY/T 88- 1988);全钾的测定则采用火焰光度法(NY/T 87- 1988);硝态氮采用连续流动分析仪法进行测定(LY/T 1228- 2015);有效磷采用钼锑抗比色法(NY/T 1121.7- 2014),其中酸性土壤使用氟化铵-盐酸作为浸提剂,碱性土壤使用碳酸氢钠作为浸提剂;速效钾则使用乙酸铵浸提,火焰光度计测定(NY/T 889- 2004/3.10)。每个土壤样品测三次取平均值记录,结果如表1所示。

表1 各采样点土壤理化性质均值(n=3)
1.4 数据分析
采用Illumina MiSeq平台对测序样本进行双端测序。高通量测试结果分析过程主要是参照Qiime2文档中名为"Atacama soil microbiome tutorial"的教程来完成(https://docs.qiime2.org/2019.1/)。原始序列通过质控、去噪、拼接及去嵌合体,形成OTU。选取代表性OTU序列,与核糖体RNA数据库(Greengenes Database 13_8版本[按99%序列相似性聚类])进行比对获得物种注释信息。
Alpha多样性以及Beta多样性的分析主要用qiime2 diversity插件完成。Alpha多样性指数用于评估样本本身的多样性程度,Beta多样性指数用于评估样本之间的微生物群落结构差异性[22]。使用了冗余分析方法(RDA)揭示微生物群落与相关环境因子之间的潜在关联。基于样本中主要微生物物种相对丰度,使用共现网络分析(Co-occurrence analysis)计算斯皮尔曼等级相关系数用于了解物种之间的关联。应用PICRUSt软件预测微生物群体可能的功能组成[23]。
2 结果与讨论
2.1 细菌群落结构分析
2.1.1测试数据分析
黑龙江表层冻土19个采样土壤中共得到原始序列785640条,随机抽取有效序列,构建Alpha多样性稀释曲线(Rarefaction Curve)(图2),各样本曲线趋于平缓,继续增加测序深度已无法再检测到大量新的OTU,表明测序结果足够反映各土壤样本中细菌的多样性。基于反距离权重插值方法,对门分类水平上每个样品的序列数目占总序列数的比例进行插值分析发现,黑龙江省表层冻土细菌序列数目占总序列数的比例自西向东呈现下降趋势。

图2 各样本Alpha多样性稀释曲线
2.1.2细菌群落组成分析
黑龙江省表层冻土检测到的细菌OTUs可划分为30个门,109个纲。209个目,326个科,512个属,598个种,其中优势菌门主要包括变形菌门(Proteobacteria)、放线菌门(Actinobacteria)、酸杆菌门(Acidobacteria)、绿弯菌门(Chloroflexi)、拟杆菌门(Bacteroidetes)、疣微菌门(Verrucomicrobia)。Proteobacteria相对丰度在49.81%至22.01%之间,采样点1处相对丰度最高。Actinobacteria相对丰度在45.33%—11.02%之间,采样点1处相对丰度最低。Acidobacteria相对丰度在17.83%—5.54%之间,Chloroflexi相对丰度在11.44%—0.73%之间,该四类菌门相对丰度之和均占各样本中细菌相对丰度的70%以上(图3),该四类优势细菌门,在P<0.01水平上,Proteobacteria与Actinobacteria、Chloroflexi极显著负相关,与Acidobacteria极显著正相关,相关系数分别为-0.748、-0.573、0.281。Actinobacteria与Acidobacteria极显著负相关,与Chloroflexi极显著正相关,相关系数分别为-0.633和0.052。Acidobacteria和Chloroflexi之间则无显著相关性。

图3 各个样本在门水平的物种相对分布
主要属水平上细菌物种相互作用网络如图4所示,圆圈代表一个物种,大小代表其相对丰度,不同的颜色代表不同的物种门分类,圆圈之间的线条代表这两个物种间的相关性显著(校正错误发现率的秩相关的P值小于0.05),线条颜色红色代表正相关,蓝色代表负相关,线条越粗,相关系数绝对值越大。大部分菌属之间的相关性均为显著正相关,只有Flavobacteriu属和Burkholderia属,Microlunatus属和Massilia属之间呈现显著负相关。对样品间具有显著性差异的属水平上物种进行ANCOM(Analysis of composition of microbiomes)分析,该方法专门用于比较物种在组间的显著性差异,其中W值越高,代表该物种在组间的差异显著性越高,clr值越高代表相对丰度差异越大。Aetherobacter在各样本之间差异最大(W=83,clr=16.58),其次为Planctmyces(W=16,clr=4.39)和Constrictibacter(W=2,clr=8.46)(图5)。

图4 主要属水平上细菌物种相互作用网络图

图5 物种属水平分类的ANCOM丰度比较结果
2.1.3细菌群落多样性分析
Alpha多样性指数用于样品中物种多样性的分析,chao1指数是用来反映物种丰富度的指标,Simpson和Shannon指数分析用于样本的物种多样性,除上面所说物种丰富度的涵义外,还指所有物种个体数目的分配的均匀程度。黑龙江省表层冻土细菌多样性chao1指数在768.6至368.0之间,其中采样点15处的指最大,其次为采样点11和18处;而采样点8处chao1多样性指数最低,其次为采样点9处。Simpson指数和Shannon指数在采样点15处值均为最大,分别为0.997和9.13,而采样点18处最小,分别为为0.986和8.18。表明采样点15处细菌丰富度较高,物种分布均匀度较好,而采样点18处虽然细菌丰富度较高,但均匀度较差。Beta多样性指数是对不同样品间的微生物群落构成进行比较分析,Bray Curtis距离是生态学上反应群落之间差异性最常用的指标,其值越小,表示这两个样品在物种多样性方面存在的差异越小。采样点1与其他样点之间Bray Curtis距离值均大于0.87,与采样点12处差异性最大,为0.99;采样点10和13处距离值为0.53,差异最小。
2.2 细菌群落功能预测
基于PICRUSt的原理对菌群代谢功能进行预测,结果如图6所示。微生物群落功能在组间差异不明显,每个样品中的菌群主要呈现20种代谢功能,其中膜转运功能基因、氨基酸代谢功能基因、碳水化合物代谢功能基因所占的相对比例较高,在每个样品中这三种功能基因均达到10%以上。其次为复制和修复功能基因、能量代谢功能基因,相对比例在5%—7%之间。其他功能基因,如信号转导、脂类代谢等相对比例均在5%以下。

图6 微生物群落功能预测组成柱形图
2.3 细菌群落与土壤理化性质相关性分析
为反映细菌与环境因子之间的关系,先进行DCA分析,最大轴的值小于4,进行RDA多元回归分析,进而得到显著影响细菌分布的环境驱动因子。门水平上(图7,图8),pH是影响细菌群落结构组成的最主要因子,与Crenarchaeota、Verrucomicrobia、Thermi呈极显著正相关,与Elusimicrobia呈极显著负相关,与Actinobacteria、Cyanobacteria呈显著正相关,与Proteobacteria、AD3呈显著负相关。其次分别为OM、C/N、WC。在P<0.001水平上,OM与Actinobacteria呈现负相关性,与Elusimicrobia呈正相关性。属水平上,pH同样是最为主要的影响因子,其次为WC、TP、OM、C/N,与这些主要环境因子具有显著相关性的菌属如图9和图10所示。追踪细菌种属关系分析发现,pH与Actinobacteria门的Arthrobacter属、Cyanobacteria门的Blastococcus属、Proteobacteria门的Chelatococcus在P<0.001水平上显著正相关相关。Kribbella属(Proteobacteria门)和Granulicella属(Actinobacteria门)则在0.001≤P<0.01水平上与pH分别呈现显著正相关和负相关。Microlunatus属(Actinobacteria门)和Microlunatus属(Proteobacteria门)则下0.01≤P<0.05水平上与pH分别呈现显著负相关和正相关。

图9 RDA排序图(属水平)

图8 门水平细菌与理化性质相关性热图
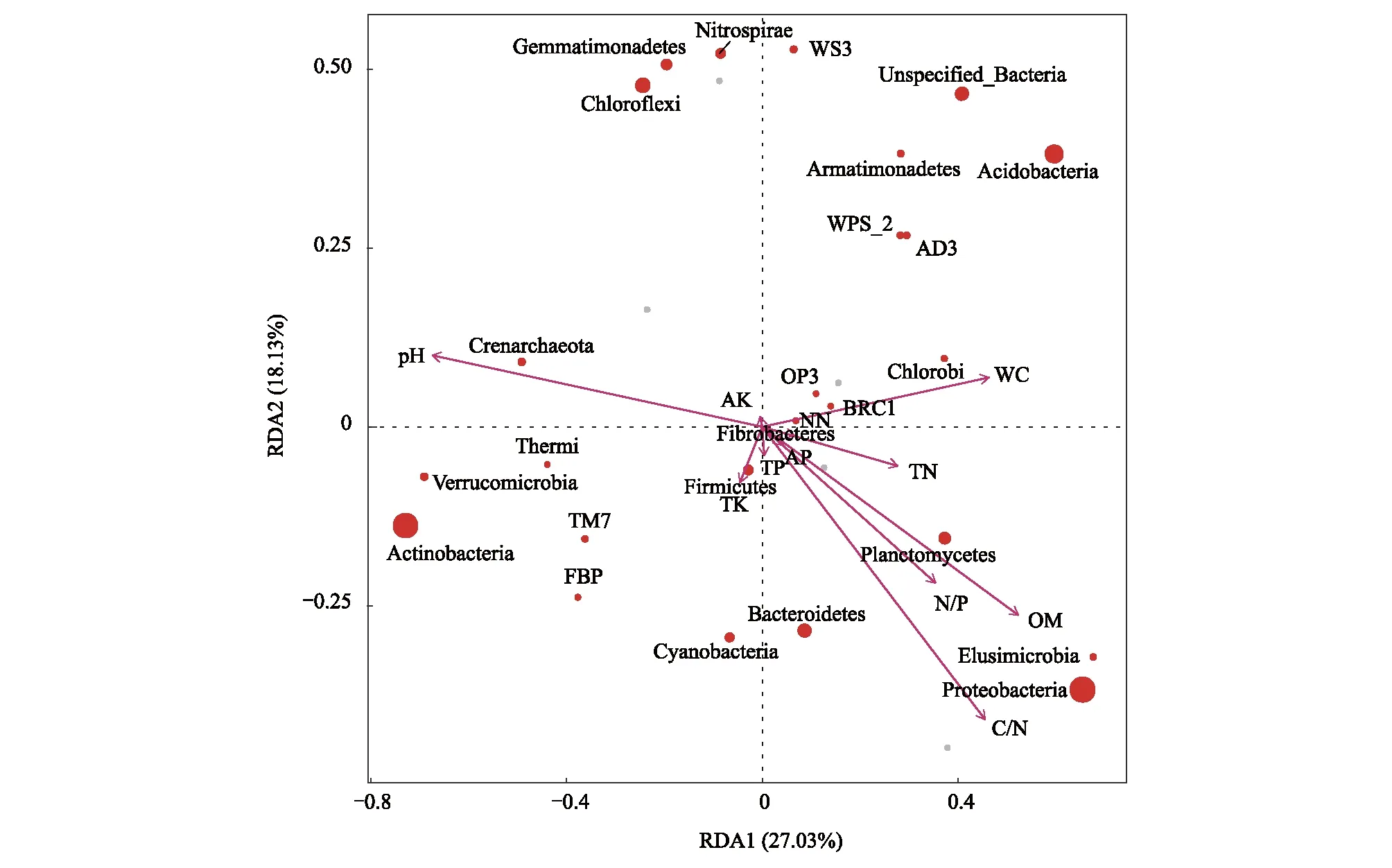
图7 RDA排序图(门水平)
2.4 利用方式对细菌群落分异影响分析
黑龙江省区域内共设置的19个采样点分属于五种利用类型土壤,Group1为森林-林地土壤(n=5),Group2为菜园-果园土壤(n=5),Group3为农田土壤(n=5),Group4为城区绿地土壤(n=5),Group5为居民区土壤(n=4),Alpha多样性以及Beta多样性在各组之间均无显著差异。选取细菌物种绝对丰度前20的菌门进行聚类分析,研究不同样品间的相似性,结果如图11所示,19个采样点被聚为6类,每类中均包括不同利用方式的土壤样品,表明利用方式不是影响黑龙江省表层冻土细菌群落结构的主要因素。
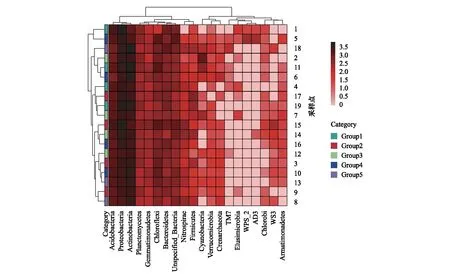
图11 不同利用类型土壤种细菌门水平的分类热图
3 讨论
土壤中的细菌群落通常由相似的优势菌门组成,包括Acidobacteria,Actinobacteria,Proteobacteria,Bacteroidetes,Verrucomicrobia和Chloroflexi等[24-25],这些菌门在冻土中也常作为优势菌门被检出[8]。王艳发等[26]对青藏高原冻土细菌群落结构研究发现,Proteobacteria、Actinobacteria作为优势菌门在冻土层中被检出。有研究表明,Actinobacteria更加适应低温环境[27],Proteobacteria在冻土在活动层具有一个很高的比例[28]。程亮等[29]研究发现,除了Actinobacteria、Proteobacteria之外,Acidobacteria、Bacteroidetes、Chloroflexi同样在青藏高原冻土中具有较高的丰富度。韩睿等[30]对青海果洛地区冻土中微生物进行测试分析发现Bacteroidetes和Proteobacteria为优势菌门。本研究中,黑龙江表层冻土优势菌门同样为以上菌门,且优势程度明显。属水平上,Aetherobacter是革兰氏阴性变形菌,广泛存在于土壤环境介质中,其富产二级代谢物,被成为自然界天然产物制造工厂,本研究中发现的Actinobacteria门的Arthrobacter属在许多冻土中被查出,如北极、南极、青藏高原、天山等,并且可作为优势菌群进行培养[11]。Planctmyces类群普遍存在于冻土环境中,且该类群经长期低温诱导,已形成较为适应该类环境的生理特性。冻土中Flavobacteriu属和Burkholderia属,Microlunatus属和Massilia属之间呈现显著负相关性则未见相关报道。
研究表明,在不同空间尺度和生态系统的冻土中,pH对细菌群落结构组成存在显著影响[14,31]。一方面是由于土壤中的细菌最适生长pH值范围较窄[32-33];另一方面是由于pH通过调节土壤养分的可利用性进而影响细菌群落结构[34]。本研究中,黑龙江表层冻土pH在5至9之间,对细菌群落结构组成影响显著,与其他人研究结果一致[35]。已有研究表明有机质含量显著影响冻土微生物群落结构[36]。碳氮是冻土中微生物的主要营养物质,微生物作为冻土碳氮循环的重要参与者,土壤碳氮含量直接对其群落的活性与分解作用产生影响[37-38],本研究中土壤C/N作为环境因素对于冻土微生物群落结构影响显著。冻土微生物数量与土壤水分含量呈现显著正相关[39],而本研究中土壤水分含量对冻土微生物群落结构的影响同样呈现显著正相关关系。李昌明[40]对青藏高原多年冻土区土壤微生物及其与环境关系的研究发现,冻土土壤理化因子中的土壤水分与养分一起控制着微生物群落丰度和组成,而冻土土壤pH值和C/N更多的调控着细菌群落的多样性,与本研究结果一致。
在对全球表层土壤微生物群落结构和功能进行研究分析发现,环境变化与细菌门水平组成上表现出强相关性,环境变量对土壤表层细菌结构及功能的影响更大[41]。土壤中细菌群落之间的相似性更多地受到土壤性质的控制(特别是土壤pH)。土壤利用[42]、管理[43]、种植模式[44]对细菌群落结构的影响也是显著的。而在本研究中,黑龙江省区域尺度范围内,表层冻土中细菌群落结构分异主要受土壤理化性质影响,而土壤利用方式对微生物群落结构影响不显著。
4 结论
黑龙江省表层冻土优势菌门主要包括Proteobacteria、Actinobacteria、Acidobacteria、Chloroflexi、Bacteroidetes、Verrucomicrobia。Aetherobacter属在各样本之间显著性差异最大,Flavobacteriu属和Burkholderia属,Microlunatus属和Massilia属之间呈现显著负相关。pH是影响细菌群落结构组成的最主要环境因子。基于细菌基因组的16S rRNA序列对菌群代谢功能进行预测发现,细菌群落主要代谢功能表现在膜转运功能、氨基酸代谢功能以及碳水化合物代谢功能三方面。冻土表层土壤理化性质的差异性决定了黑龙江省区域尺度范围内土壤细菌群落结构的分异,而本研究中涉及的土壤利用方式对微生物群落结构影响不显著。
猜你喜欢
海洋石油(2021年3期)2021-11-05
河北环境工程学院学报(2021年1期)2021-03-19
中国比较医学杂志(2020年4期)2020-05-26
小哥白尼(趣味科学)(2020年7期)2020-05-22
水生生物学报(2019年4期)2019-07-20
生物安全学报(2019年3期)2019-02-15
川北医学院学报(2019年6期)2019-02-10
小哥白尼(趣味科学)(2018年9期)2018-12-18
时代英语·高三(2014年5期)2014-08-26
组合机床与自动化加工技术(2014年12期)2014-03-01
