
Genomes of major fishes in world fisheries and aquaculture:Status,application and perspective
2020-08-08 07:50GuoqingLuMingkunLuo
Aquaculture and Fisheries 2020年4期
Guoqing Lu,Mingkun Luo
a Department of Biology,University of Nebraska at Omaha,Omaha,68182,USA
b Wuxi Fisheries College,Nanjing Agricultural University,Jiangsu,Wuxi,214081,China
A R T I C L E I N F O
Keywords:
Fish genomics
World aquaculture
Fisheries management
Genomic selection
Intelligent system
A B S T R A C T
Capture fisheries and aquaculture provide a significant amount of high-quality protein to human beings and thus play an essential role in ending global hunger and malnutrition.The availability of tens of hundreds of fish genomes and the advances of genomics have allowed addressing many challenging issues such as overfishing and germplasm degradation faced by fisheries and aquaculture.In this review,we describe the current status of genomics in fisheries and aquaculture,with an emphasis on 14 species of fish that are considerably important to global fisheries and aquaculture,in the context of genome sequencing and assembly,annotation,GC contents,and repeats.The majority of these genomes are assembled at the chromosome level and annotated with proteins and pathways,with functional relevance to fisheries and aquaculture,such as environmental adaptation and phenotypic variation.We summarize potential genomic applications in fisheries and aquaculture that are related to assessment and use of genetic resources,disease resistance,growth and development,sexual determination,and fisheries management.Although much progress has been achieved in genomic application to fisheries and aquaculture,the full potential remains to be explored and reaped.We discuss the challenges and perspectives of genomics in translational aquaculture and fisheries,which include genome assembly and annotation,genomic selection and breeding,genomics in fisheries management,and integrated artificial intelligence systems.In the coming decades,we anticipate the applications of genomic techniques such as genome editing and genomic selection,along with the use of emerging intelligence systems,in aquaculture and fisheries will contribute significantly to genetic improvements of farmed fish and sustainable exploitation of fishery resources,which consequently lead to eradicating global poverty by 2030,an ambitious goal set by the United Nations.
1.Introduction
Capture fisheries and aquaculture contribute over 15 percent of animal proteins in human consumption and thus play an essential role in eradicating poverty and achieving sustainable development worldwide by 2030,an agenda set by the United Nations(Bernatchez et al.,2017;FAO,2018).Remarkable success has been achieved in world fisheries and aquaculture during the past decades.According to the 2018 statement by the Food and Agricultural Organization of the United Nations(FAO),the average annual increase between 1961 and 2016 in global food fish consumption(3.2 percent)outpaced global population growth(1.6 percent)and exceeded that of meat from all terrestrial animals combined(2.8 percent)(FAO,2018).The long-term sustainability of fisheries and aquaculture,however,faces many challenges,including overfishing,climate change,germplasm degradation,and diseases(Bernatchez et al.,2017;FAO,2018;Yue & Wang,2017).The development of sequencing technologies and the advances of genomics are instrumental in addressing some of these challenges and can benefit sustainable fisheries and aquaculture(Bernatchez et al.,2017;Canário,2019;Yue & Wang,2017;Liu,2010).
The first fish with its whole genome sequenced was Japanese pufferfishFugu rubripes(Aparicio et al.,2002).This food fish has been commercially cultured in Japan and China(Froese&Pauly,2010).With the development and advancement of massive parallel sequencing technologies originating around 2005(Heather & Chain,2016),over two hundred fish genomes have been sequenced and made available in public repositories.For example,as of Dec 21,2019,some 270 assembled fish genomes were available in the NCBI Genome(Table S1).These genomic resources promote not only basic sciences such as comparative genomics,evolution,and systematics but also applied practices in aquaculture and fisheries(Bian et al.,2019;Hughes et al.,2018;MacKenzie&Jentoft,2016;Roest Crollius&Weissenbach,2005).The Genome 10 k Project launched in 2009 by a consortium of biologists and genome scientists targets at sequencing the complete genomes of 10,000 vertebrate species,including some 4000 fish species to understand vertebrate evolution and save many endangered species(Bernardi et al.,2012;Haussler et al.,2009;Koepfli,Paten,O'Brien,&Scientists,2015).The Earth BioGenome Project(EBP),a moonshot for biology,aims at sequencing,cataloging,and characterizing the genomes of all of the Earth's eukaryotic biodiversity over ten years(Lewin et al.,2018).Regardless,all known species of fish(over 34,000 species recorded in FishBase,http://fishbase.org)will soon have their genomes sequenced.The subsequent challenges are how to make use of these genomic data and transform genomic knowledge into fisheries and aquaculture practices,such as genetic resources management and selective breeding.
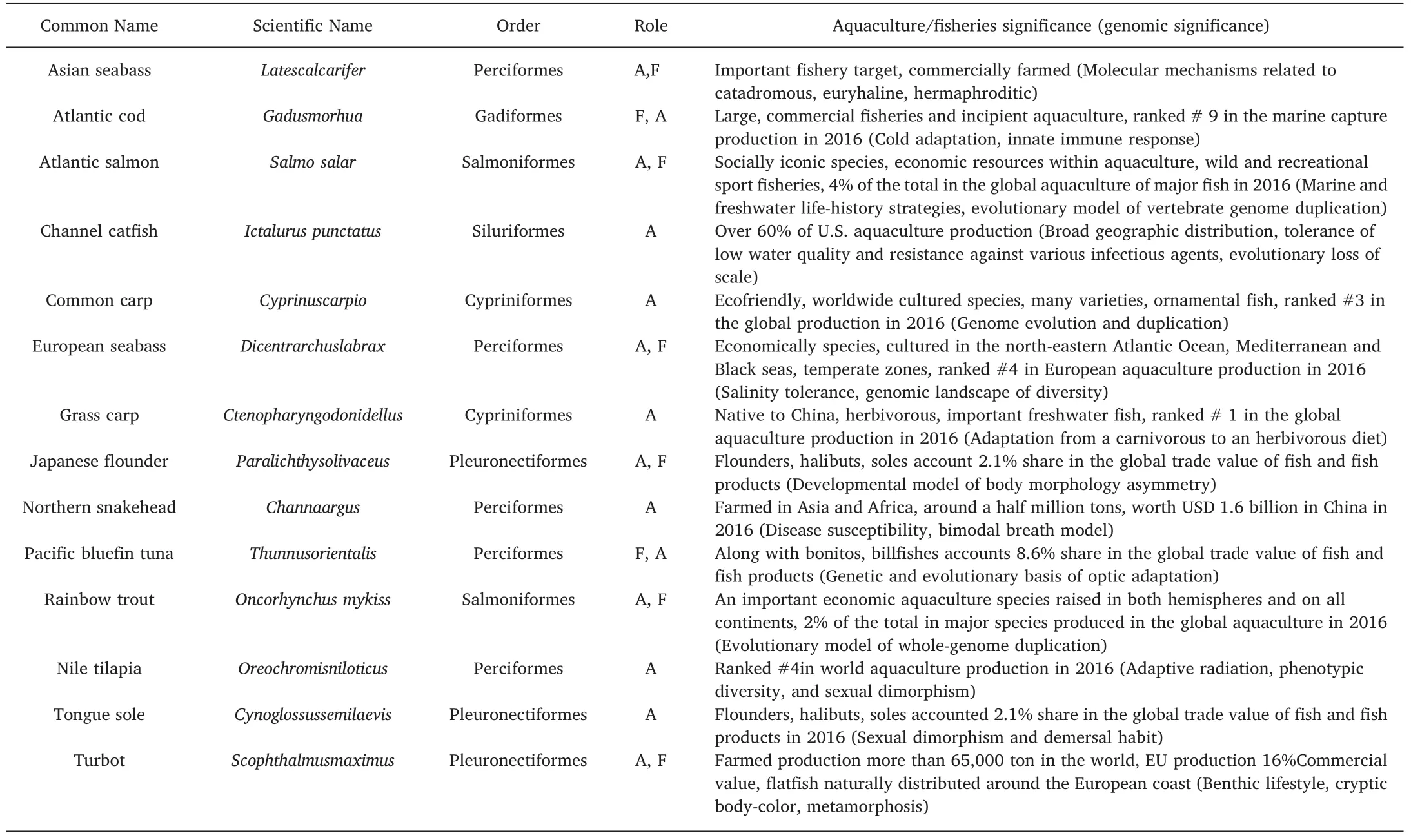
Table 1Major fishes in world fisheries and aquaculture discussed in this review and their genomic significance.
Aquaculture genomics identifies the genetic basis of performance and production traits and utilizes such information for selective breeding programs.It has been discussed in review articles(Abdelrahman et al.,2017;Yue & Wang,2017),conference reports(Bernatchez et al.,2017;Shen & Yue,2019),and reference books(Liu,2010;MacKenzie & Jentoft,2016).Of note,Yue and Wang(2017)reviewed the status of genome sequencing and its various applications such as breeding,diseases,sex determination and maturation in fish,oysters and mitten crab.Abdelrahman et al.(2017)discussed current status,challenges,and priorities for future research in aquaculture genomics,genetics,and breeding in the United States.Fisheries genomics,an emerging field that applies genomic tools to solve questions related to fisheries management,has been discussed less frequently as compared to aquaculture genomics.The use of genomic tools in fisheries can detect adaptive diversity in response to fishing effort,reveal stock identity and structure,and estimate abundance and spawning stock biomass(Casey,Jardim,& Martinsohn,2016;Valenzuela-Quiñonez,2016).However,when compared to agriculture,aquaculture and fisheries appear to fall behind in applying genomics in selective breeding and resources management(Bernatchez et al.,2017).In this review,we intend to fill this gap by introducing sequencing technologies and genomics and describing genomic findings of major fish in world fisheries and aquaculture concerning genome structure,organization,and function.Furthermore,we present major applications of fish genomics in aquaculture and fisheries and discuss future perspectives in genomic selection and fisheries management.
Considering the contribution to world fisheries and aquaculture production,we are determined to focus on 14 major fish species in aquaculture and fisheries(Table 1;FAO,2018).These species are from diverse groups,including Cypriniformes(grass carp and common carp),Gadiformes(Atlantic cod),Perciformes(European seabass,Nile tilapia,Asian seabass,Pacific bluefin tuna,and northern snakehead),Pleuronectiformes(tongue sole,turbot,and Japanese flounder)and Salmoniformes(rainbow trout and Atlantic salmon).The grass carp,common carp,and Nile tilapia are among the most important fish species in world aquaculture.European seabass,Asian seabass,Japanese Flounder,rainbow trout,and Atlantic salmon are vital species in aquaculture as well as fisheries.Atlantic cod and Pacific bluefin tuna are crucial species in marine fisheries.
2.Brief history of genomics and genome sequencing technologies
A genome is the entire collection of all functional and nonfunctional DNA sequences in an organism,and thus provides insights into what makes a species unique in aquaculture and/or fisheries.Genomics became a discipline around 1987 and originated with the advance of molecular genetics and the development and application of various biotechnologies.The DNA's double-helix structure discovered by Watson and Crick(1953)symbolized the new era of molecular biology.The DNA sequencing technologies were invented in the 1970s,with Sanger sequencing still widely used(Sanger,Nicklen,&Coulson,1977).The subsequent improvements of the Sanger's method led to the development of ABI DNA sequencing systems(MacKenzie & Jentoft,2016).It was the main platform used for sequencing the human genome and the genomes of model fish organisms,including torafugu(Aparicio et al.,2002),spotted green pufferfishTetraodon nigroviridis(Jaillon et al.,2004),medaka,i.e.,Japanese rice fishOryziaslatipes(Kasahara et al.,2007),and zebrafishDanio rerio(Howe et al.,2013).
DNA sequencing technologies have advanced dramatically and rapidly since the human draft genome was published(Lander et al.,2001).The Sanger method produces high-quality DNA sequences but has drawbacks of low throughput and high cost(Heather & Chain,2016).The next-generation sequencing(NGS)technologies were developed to resolve the shortcomings of first-generation sequencing technologies(Sanger sequencing)and allow sequencing tens of millions of nucleotides in a single run(Heather & Chain,2016;Shendure et al.,2017).The second-generation sequencing methods include pyrosequencing(454 Life Sciences)that was introduced in 2005(Margulies et al.,2005),bridge amplification-based sequencing(Solexa,acquired by Illumina)(Ruparel et al.,2005;Seo et al.,2005),and sequencing by oligonucleotide ligation and detection(SOLiD)(Life Technologies,acquired by Thermo Fisher Scientific)(Heather & Chain,2016).The second-generation sequencing technologies have resulted in the oftdescribed“genomics revolution”,with no exception in fisheries and aquaculture.
The main disadvantages of the second-generation sequencing methods are short sequencing reads and significant time per sequencing run,which leads to the birth of third-generation sequencing technologies.These latest sequencing technologies,represented by Oxford Nanopore Technologies(ONT)or SMRT Pacific Biosciences(PacBio)sequencing platforms,can produce long reads and require relatively short running time(Astier,Braha,&Bayley,2006;Eid et al.,2009).The major limitation of these third-generation sequencing technologies is their accuracy.However,high sequencing coverage could overcome this issue.The combined use of second-and third-generation sequencing technologies,such as Illumina and PacBio appear to be a practical approach forde novosequencing non-model organisms(J Wang et al.,2019).The emergence of fourth-generation sequencing technologies(e.g.,Hi-C technology),such asin situsequencing,combines traditional imaging analysis techniques and the NGS technologies to sequencing nucleic acids directly in cells and tissue(Ke,Mignardi,Hauling,&Nilsson,2016;Mignardi&Nilsson,2014).The combination of Illumina,PacBio,and Hi-C technologies has been applied to construct the chromosomal-level genome assembly of yellow catfish,an economically important freshwater aquaculture fish species in Asia,especially in Southern China(Gong et al.,2018).

Table 2Genome assembly and gene annotation of major fishes in world fisheries and aquaculture.
3.The genomes of major fish in world aquaculture and fisheries
3.1.Genome sequencing and assembly
Sequencing was conductedde novousing next-generation sequencing technologies such as 454 and Illumina and PacBio(Table 2).Most sequencing projects were advanced based upon previous research on linkage mapping and quantitative traits linkage(QTL),which enabled chromosome-level genome assemblies(Conte,Gammerdinger,Bartie,Penman,& Kocher,2017;Wang et al.,2015;Xu et al.,2014).The estimated genome sizes ranged from 544 Mb in turbot(Figueras et al.,2016),a flatfish to 2.97 Gb in Atlantic salmon(Lien et al.,2016).Among the 14 major species,brown trout,a tetraploid species in Salmonidae,possessed the second-largest genome of 2.18 Gb(Berthelot et al.,2014).Common carp,which is also a tetraploid species,had a genome of 1.83 Gb(Xu et al.,2014).The flatfish species and northern snakehead had relatively small genomes(Figueras et al.,2016;Xu et al.,2017).The genome size of grass carp was approximately 1.07 Gb for a female and 0.9 Gb for a male(Wang et al.,2015).The scaffold N50,an indicator of the genome completeness,ranged from 137 kb in Pacific bluefin tuna to 7.7 Mb in channel catfish(Chen et al.,2016;Nakamura et al.,2013).Grass carp genome had the second-largest N50 of 6.5 Mb,comprising 114 scaffolds(Wang et al.,2015).Only four species had the scaffold N50 of genomic sequences<1 Mb(Table 2).It is worth noting that multiple factors,such as the coverage and assembly software,may affect the estimated scaffold N50 values.
3.2.Genome annotation
One of the main tasks in genome annotation is to identify proteincoding genes.The predicted protein-coding genes in the genome of these 14 fish species ranged from 19,877 in Northern snakehead to 52,610 in common carp(Table 2).The number of predicted protein-coding genes almost doubled in common carp compared to zebrafishD.rerio,demonstrating that the tetraploid genome of common carp likely retained a large portion of duplicated genes since the latest whole-genome duplication(Xu et al.,2019;Xu et al.,2014).The Atlantic salmon genome appeared to have experienced a crucial post-Ss4R(salmonid specific fourth vertebrate whole-genome duplication)rediploidization process that resulted in large genomic reorganizations and was demonstrated by the bursts of transposon-mediated repeat expansions(Lien et al.,2016).In rainbow trout,two ancestral sub-genomes remained extremely collinear after 100 million years.Interestingly,only 22,184 protein-coding genes were predicted in the rainbow trout genome,which could be attributed to the loss of half of the duplicated genes mostly through pseudogenization(Berthelot et al.,2014).
Functional genome annotation allowed the prediction of genes associated with biological traits that are of significant importance in fisheries and aquaculture(Table 2).The Atlantic cod genome possessed highly expanded MHCI genes and Toll-like receptor(TLR)families,indicating its unique immune system in response to infectious disease(Table 3)(Star et al.,2011).In tongue sole,the Gene Ontology(GO)analysis of differentially expressed genes between individuals before and after metamorphosis unveiled metamorphic transition and environmental cues linked to a benthic lifestyle(Shao et al.,2017).In grass carp,the KEGG(Kyoto Encyclopedia of Genes and Genomes)pathway analysis identified genes associated with adaptation from a carnivorous to an herbivorous diet that was likely involved in transcription activation of the mevalonate pathway and steroid biosynthesis(Wang et al.,2015).The Japanese flounder has the most extreme asymmetric body morphology,and one eye migrates to the contralateral side of the skull.Its genome was enriched with gene families in the GO categories of collagen,microtubules,regulation of appetite,and protein polymerization and with KEGG pathways related to cell proliferation and apoptosis,carbohydrate metabolism,and cytokines and immune responses(Shao et al.,2017).In common carp,the genomic comparison between Hebao and Songpu strains located 205 regions with the highest genetic diversity that contained 326 candidate genes(Xu et al.,2014).The GO and KEGG analyses of the common carp genome suggested a significant portion of these genes associated with epithelial morphogenesis,pigmentation,and immune response,including growth factor signaling and other functional pathways(Xu et al.,2014).Collectively,the annotation of fish genomes has revealed numerous molecular mechanisms underlying phenotypic traits linked to aquaculture(Table 3).
3.3.Genomic composition-GC contents
The base composition,although highly variable among species,is an important genomic feature related to genomic function and species ecology(Smarda et al.,2014).In teleost,the GC content was found to be higher in seawater than freshwater fish and higher in migratory than non-migratory species,suggesting a correlation between genomic GC contents and living environments(Tarallo et al.,2016).Among the 14 species,GC content ranged from 31.5% in channel catfish to 45.8% in Atlantic cod(Table 2).In European seabass,a lower percentage of GC was observed in noncoding regions(39.6%)compared with proteincoding regions(52.6%),and the GC content in the third codon positions was 60.7%.This observation suggests possible selection effects on codon usage or biased gene conversion in coding regions compared to noncoding regions(Duret & Galtier,2009).An inverse relationship between the genome size and GC contents reported in previous studies was found insignificant based on the genomic data of these 14 species,indicating more genomic data are needed for the validation of this assertion(Howe et al.,2013;Tine et al.,2014).
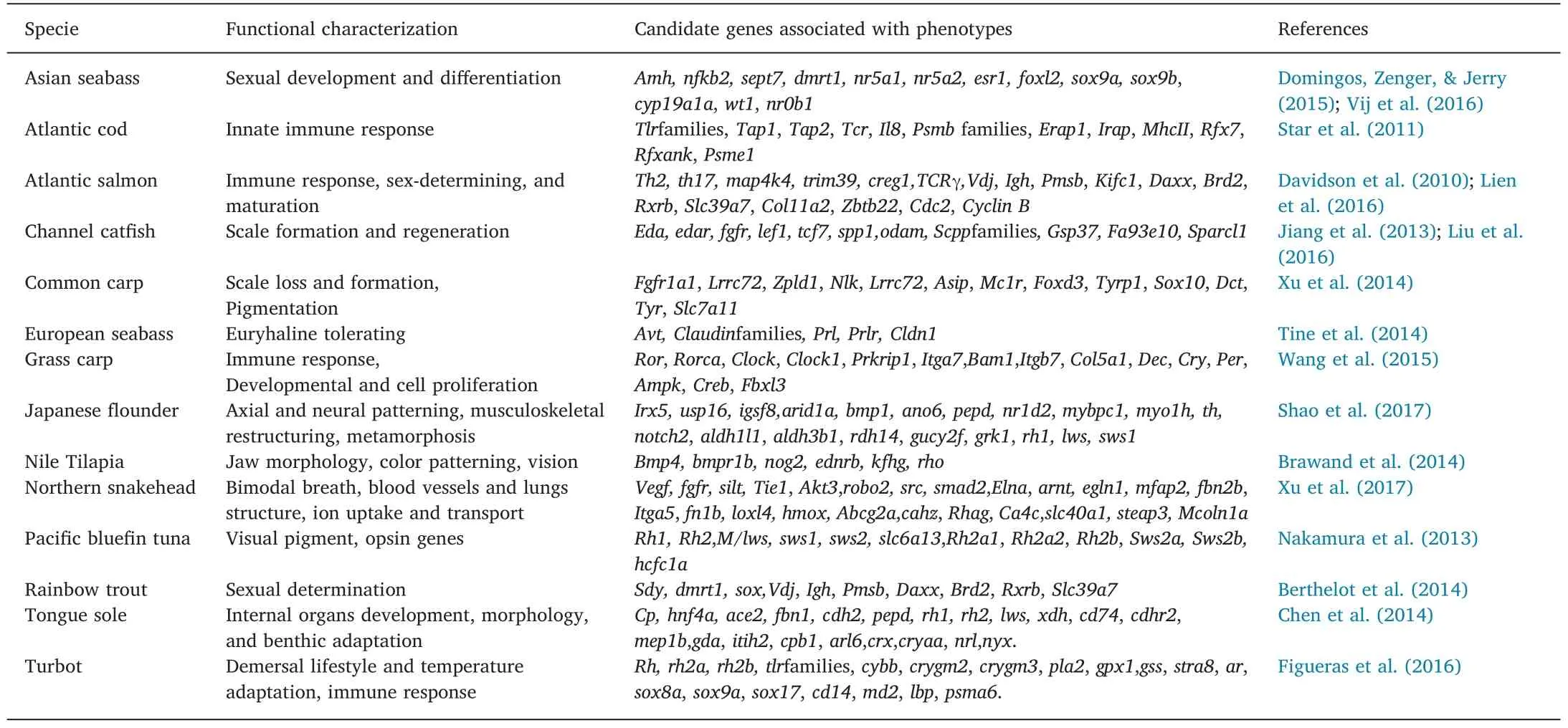
Table 3Genes associated with functional characterization important in aquaculture and fisheries.
3.4.Genomic structure-repeats
Repetitive elements(repeated sequences or repeats)occur in multiple copies and account for a significant portion of the genome.A comparative genomic analysis of 52 fish species found the proportion of repetitive elements was positively correlated with genome sizes,and certain repetitive element categories were enriched in marine versus freshwater species(Yuan et al.,2018).Pleuronectiformes flatfish possessed 6.0-9.0% repeats,which is comparable to takifugu(7.1%)(Aparicio et al.,2002),Tetraodon(5.7%)(Jaillon et al.,2004),and stickleback(13.48%)(Jones et al.,2012).Repetitive elements accounted for 58-60% in the salmon genome,which is likely associated with its genome duplication(Lien et al.,2016).Repetitive elements in common carp and rainbow trout were found to be relatively high,which could also be explained by genome duplication(Berthelot et al.,2014;Xu et al.,2014).The high content of repetitive elements in the genome could increase the birth of new genes;however,it could also result in genomic instability due to possible abnormal recombination or splicing(Yuan et al.,2018).The definite role of repetitive elements in the genomic function of fish remains to be explored.
4.Genomic applications in fisheries and aquaculture
Genomics allows identifying genetic basis associated with aquaculture traits and detecting genetic variations caused by environmental changes and thus has many applications in fisheries and aquaculture.These applications include accurate identification of fish stocks for capture fisheries management,conservation of fish genetic resources,genome selection for genetic enhancement,disease resistance,and sex determination and control.We present below some of the most important genomic applications to fisheries and aquaculture.
4.1.Assessment and use of genetic resources
Common carp is not only the most representative carp species in aquaculture but also the most popular outdoor ornamental fish with its distinctive color and scale patterns(Xu et al.,2014).The whole-genome sequencing resulted in a high-quality genome assembly of the common carp Songpu strain.The genomic resequencing of 33 individual fish representing major domesticated strains and populations revealed phylogenetic relations and population structure of worldwide populations and identified 19 million candidate SNPs and 1.7 million small insertion-deletions.The genomic comparison,along with transcriptomic analysis,identified genetic loci likely associated with scale patterns and skin color(Table 3).The draft genome of the common carp thus provides an important genomic resource to study the genetic basis of economically important traits and to facilitate genome-based genetic breeding technologies in common carp aquaculture(Xu et al.,2014).
Cichlid fishes are known for diverse and replicated adaptive radiations in the Great Lakes of East Africa.The genome sequencing of African cichlids in five different lineages uncovered several molecular mechanisms potentially involved in their phenotypic diversity,including excessive gene duplications,abundant non-coding element divergence,accelerated coding sequence evolution,expression divergence associated with transposable element insertions,and regulation by novel microRNAs(Brawand et al.,2014).The genomic analysis of chromosome-scale assemblies found intra-chromosomal structural differences(~2-28 Mb)among species are common,while inter-chromosomal differences are rare(<10 Mb).In addition,the sequence analysis of 60 individuals representing six closely related species from Lake Victoria showed genome-wide diversifying selection on coding and regulatory variants(Brawand et al.,2014).
The European seabass,native to the north-eastern Atlantic Ocean and throughout the Mediterranean and Black seas,is an economically important species(Tine et al.,2014).Its natural stocks are subject to intensive exploitation,which consequentially raises concerns about conservation and management.The high-quality chromosome-scale assembly of the European seabass genome allowed identifying expansions of gene families linked to ion and water regulation,revealing possible adaptation to variation in salinity in this coastal fish.The sequence mapping of genome-wide variations between Atlantic and Mediterranean populations showed that the genomic landscape of diversity within and differentiation between lineages was contributed by variation in local recombination rates and diverse genomic introgression following the post-glacial secondary contact(Tine et al.,2014).
4.2.Disease resistance
Disease control represents one of the main challenges in aquaculture.The immune-related molecular understanding through analysis and comparison of genomes is essential for the development of effective therapeutics,including vaccines and drugs,to control diseases.Specific families of pattern recognition receptors are responsible for detecting microbial pathogens and generating innate immune responses.Pathogen recognition by Toll-like receptors(TLRs)causes rapid activation of innate immunity by inducing the production of proinflammatory cytokines and upregulation of costimulatory molecules.In Atlantic cod,most genes involved in vertebrate innate immune response were found,except for major histocompatibility complex(MHC)II genes(Table 3;Star et al.,2011).The MHC II is a conserved feature in the adaptive immune system of jawed vertebrates.None of the MHC isoforms,trafficking chaperone II,or the MHC-II-interacting protein CD4 was recovered in the Atlantic cod genome,suggesting an alternative immune response mechanism may exist.The marked expansion of MHCI genes and unusual TLR composition indicate a shift of its immune system in handling microbial pathogens.Further analysis of draft genomes of 66 teleost fish confirmed the loss of MHC II and the expansion of MHC I gene clusters in Gadiformes,an order that includes Atlantic cod(Malmstrom et al.,2016;Malmstrom,Matschiner,Torresen,Jakobsen,& Jentoft,2017).
The genome sequencing and assembly of turbot recovered a complete repertoire of bacterial and viral TLRs except for a few genes,including TLR4,an important gene in the immune pathway(Figueras et al.,2016).The missing of TLR4 in turbot is consistent with the finding in some teleost fish,indicating the variation of the TLR families among fish species.In Atlantic salmon,the infectious pancreatic necrosis QTL has been identified and successfully applied in aquaculture practice(Houston et al.,2008).The large yellow croaker,Larimichthys crocea,is an economically important marine fish endemic to China.Its natural resource has been overexploited,and the farmed stocks are vulnerable to various pathogens.The genomic analysis of a wild large yellow croaker showed that fast-evolving genes under positive selection were significantly enriched in pathways related to innate immunity(Wu et al.,2014).The identification of innate immunity-related gene family expansions suggests a well-developed innate immune system in the large yellow croaker(Wu et al.,2014).
4.3.Growth and development
Improving growth performances is a central goal of breeding programs in fish;however,growth is under complex genetic control and can be impacted by environmental,metabolic,and physiological factors(De-Santis & Jerry,2007;Johnston,Bower,& Macqueen,2011).In turbot,growth-associated QTL markers and 208 selected candidate genes from fish and vertebrates were mapped onto the genome,and most of them were predictively located on the turbot genetic map(De-Santis & Jerry,2007).Remarkably,a number of genes previously used for growth-assisted selection in livestock and other aquaculture species were found within a major Fulton's factor-QTL at LG16(Figueras et al.,2016).
The formation and development of morphological traits are complex and often controlled by genetic and environmental factors and their interactions.Common carp(Cyprinus carpio)is a genetically diverse species and has emerged across a broad spectrum of ecological settings.The genome sequence of common carp showed that deletions in coding regions offgfr1a1gene(fibroblast growth factor receptor 1 a1)resulted in a reduced-scale phenotype(Table 3)(Xu et al.,2014).Theslc7a11gene(solute carrier family 7 member 11),the plasma membrane cystine/glutamate exchanger that transports cystine into melanocytes to synthesize pheomelanin(yellow to red pigment),is significantly more upregulated in the skin of Hebao strain than that of Songpu strain(Xu et al.,2014).
The comparative analysis of genomes and transcriptomes of scaled and scaleless fish and the scale regeneration experiments showed that five genes,found in zebrafish and involved in scale development,were all present and expressed in channel catfish(Table 3)(Chen et al.,2016).The comparison of gene structure and expression showed the lack of certain secretory calcium-binding phosphoproteins might account for the evolutionary loss of scales in catfish(Chen et al.,2016).This work demonstrated the power of the comparative subtraction of candidate genes for traits of structural significance.
Grass carp is one of the most important species in global aquaculture.The sequencing of the grass carp genome did not recover any genes coding for cellulose degradation enzymes.However,during the food habit transition period from a carnivorous to an herbivorous diet,differential expression genes were significantly enriched in pathways associated with the circadian rhythm in guts and mevalonate pathway and steroid biosynthesis pathway in liver(Table 3)(Wang et al.,2015).The authors speculated that the intake of high-intensity food might warrant sufficient nutrients for grass carp to maintain its rapid growth(Wang et al.,2015).The grass carp genome provided important support for the discovery of genes related to important economic traits of herbivorous fish and the genetic improvement of new stocks.
The flatfish change its body shape from symmetric to asymmetric during the development at the metamorphosis stage.The genomic comparison of two flatfish(tongue sole and Japanese flounder)and transcriptomic analyses revealed thyroid hormone,retinoic acid singling,and phototransduction pathways most likely involved in metamorphosis in flounder(Shao et al.,2017).The retinoic acid was found to play a vital role in the formation of asymmetric pigmentation and the modulation of eye migration.The expression of visual opsins in skin resulted in retinoic acid gradients that may underlie the generation of asymmetry(Table 3;(Shao et al.,2017).
4.4.Sex determination
The biological sex in fish is a complex trait and shows a high evolutionary turnover closely related to genetic,environmental,and social factors(Martinez et al.,2014;Mei&Gui,2015).Due to the significance of sex in reproduction,growth,and product quality,the ability to control sex is considered one of the most important measures in fish selective breeding(Budd,Banh,Domingos,& Jerry,2015).The wholegenome sequencing,along with transcriptome sequencing and molecular cytogenetic analysis,has allowed the identification of many sexdetermining genes and mechanisms in fishes(Li et al.,2016;Reichwald et al.,2015;Rondeau et al.,2013).For instance,turbot females grow much faster than males,and the turbot genome has facilitated identifying sex-related genes for marker-assisted selection(Figueras et al.,2016).
The diversity of fish sex-determining genes arises not only from sexual regulatory networks but also fromde novoevolution of other genes(Li & Gui,2018).Asian seabass is hermaphroditic,with its sex transforming from male to female following maturation.The genomic analysis showed that specific duplications of genes such as anti-mullerian hormone(amh)and nuclear factor kb2(nfkb2)were enriched for functions related to gonad development.Both genes were reported to play an essential role in gonad transformation in zebrafish and exhibited differential expression levels between male and female gonads(Table 3;Vij et al.,2016).
The salmonid master sex-determining gene(i.e.,sdy)arose from the duplication of the autosomal interferon regulatory factor 9(irf9).It can prevent the female differentiation by interacting and blocking the action of a critical ovarian differentiation factor in rainbow trout(Berthelot et al.,2014).Overexpression ofsdyin rainbow trout was found to induce testicular differentiation,whereassdyinactivation led to ovarian differentiation(Yano et al.,2012).In Atlantic cod,six different regions on five different linkage groups in the genome were associated with sex(Star et al.,2011).One identified region on LG11 was used to predict gender with consistently high accuracy,which was most likely the sex-determining regions in Atlantic cod.The whole-genome sequencing thus proved to be a valuable strategy for detecting small regions associated with sex in Atlantic cod.The results highlight evolutionary flexibility in genomic architecture underlying teleost sex-determination.
The genome sequencing of one male(ZZ)and one female(ZW)tongue sole found that the sex chromosomes of this fish were derived from ancestral vertebrate proto chromosomes as the avian W and Z chromosomes did(Chen et al.,2014).Remarkably,thedmrt1gene on the Z chromosome,which is the male-determining gene in birds,showed convergent evolution of features that were compatible with a similar function in tongue sole.The comparison of sex chromosomes among tongue sole,birds,and mammals uncovered events occurred during the early phase of sex-chromosome evolution.Massive gene loss was found to occur in the wake of sex-chromosome‘birth’,which supports the hypothesis of heterogametic sex-chromosome decay(Chen et al.,2014).
Tilapias are among the most important farmed fishes,and its aquaculture production continues to increase globally.An essential aspect of commercial production is the control of sexual differentiation.Male tilapias grow to market-size earlier than females.Females start to reproduce at a smaller size,which often results in production ponds filled with small fish.It is,therefore,advantageous to grow out only male fish(Conte et al.,2017).The new assembly of Nile tilapia identified the long-range structure of both a~9 Mb XY sex-determination region on LG1 inO.niloticusand a~50 Mb WZ sex determination region on LG3 in the related speciesO.aureus(Conte et al.,2017).This study provides an example that high-quality genome assemblies are critical for the identification of sex-determining genes in fish species.
4.5.Fisheries management
The whole-genome sequencing of Asian seabass and resequencing of population samples from the tropical Asian Pacific region resolved its population structure and facilitated the analysis of genetic diversity(Vij et al.,2016).Three distinct phylogenetic groups,Indian region,SE Asia/Philippines,and Australia/Papua New Guinea,were identified,likely arising from allopatric origins,with evident admixture in the SE Asian population.The assessment of genomic diversity among populations across the native range allowed the identification of over 5.6 million SNPs.This genomic resource is very important in the development of genomics-based assays that will benefit Asian seabass aquaculture(Vij et al.,2016).
The Atlantic cod is one of the fishery resources most studied with molecular markers(Valenzuela-Quiñonez,2016).The use of genomic tools allowed the discovery of adaptive diversity among populations in Atlantic cod,while low levels of population structure were detected using conventional markers(Bradbury et al.,2010,2013).In contrast to neutral markers,outlier loci showed different levels of population structure and revealed parallel evolution related to temperature clines on both sides of the Atlantic Ocean(Bradbury et al.,2010).Patterns of neutral markers were related to geographic barriers between east and west Atlantic,while the inclusion of outlier loci revealed a finer-scaled structure,detecting north-south discontinuities on both sides of the Atlantic(Bradbury et al.,2013).The FishPopTrace,an international program of the European Union for monitoring,control,and surveillance of fisheries products,uses thousands of SNP markers for tracing relevant commercial fishes,including Atlantic cod(Martinsohn,Ogden,&Consortium,2009).Outlier SNP panels with the minimum number of markers for the assignment of original populations resulted in 93-100% of correct assignments among species to their populations of origin(Nielsen et al.,2012).Identifying outlier SNPs in genomic regions under selection appears to be a powerful approach for diagnostic analysis of population origin and fisheries enforcement.
4.6.Environmental adaptation
The Northern snakehead,an economically important freshwater fish native to East Asia,can breathe air with gills and migrate short distances over land.The genome sequencing of the Northern snakehead identified several essential genes in the angiogenesis pathway(related to gas exchange)and detected ten expressed genes in the iron pathway,indicating maintaining ion uptake and transport balance is an essential function of the gill(Xu et al.,2017).The turbot fish are adapted to a benthic lifestyle.They live in shallow waters during larval and juvenile stages,and afterward,move to the deep waters upon reaching adulthood.The whole-genome sequencing of turbot showed that the most key biochemical response of poikilothermic organisms to environmental cooling was to increase their unsaturated fatty acids of both membrane and depot lipids(Figueras et al.,2016).The membrane polyunsaturated fatty acids(PUFA)content of marine fish is usually higher than other vertebrate taxa.The genomic analysis in turbot identified five copies of phospholipase A2(Pla2)and two copies of dependent-glutathione peroxidase(Gpx)genes that were required to prevent oxidative damage.Positive selection was detected in threepla2genes(turbot2,turbot3,and turbot4)(Table 3)(Figueras et al.,2016).The only fish species with morepla2copies than turbot was the Atlantic cod,which inhabits very cold waters,while all other fish showed two copies at most.Two copies of the glutathione synthetase genes(gss),an enzyme involved in glutathione synthesis,were identified in turbot in which turbot was found to be the only fish species with twogsscopies(Figueras et al.,2016).
5.Perspectives
5.1.Genome sequencing and annotation
The aquaculture and fisheries genomics have made significant progress during the past decade since the first key fisheries species,Atlantic cod,was sequenced in 2011(Star et al.,2011).More genomes are available for the study of molecular mechanisms involved in the aforementioned genomic applications.The genomic data in public repositories should,however,be used with caution because the quality of draft genomes varies greatly in different fish projects,which can be attributed to many factors such as the sequencing technologies and software tools used(Table 2).The majority of published genomes were sequenced using second-generation sequencing technologies,which produce short reads that are challenging in assembly.The use of different sequencing technologies(including fourth-generation Hi-C)can fill this gap and produce chromosome-level,high-quality genomes(Yue& Wang,2017).
The completeness of whole-genome sequencing refers to the percentage of the genome sequenced(FAO,forthcoming).We noticed multiple assemblies submitted by different research groups existed in several fish species(Table S1);it is thus imperative to combine sequencing reads and come up with a reference genome for each species.Regarding the assessment of genome assembly completeness,we often use the core gene sets such as CEGMA(Core eukaryotic genes dataset)(Parra,Bradnam,&Korf,2007)or universal single-copy orthologs such as BUSCO(Simao,Waterhouse,Ioannidis,Kriventseva,& Zdobnov,2015).However,CEGMA or BUSCO does not have a benchmark dataset dedicated to fish.It is thus valuable to establish a core set of universal single-copy genes in fish,which can be achieved using bioinformatics tools such as OrthoMCL(Li,Stoeckert,& Roos,2003),BUSCO(Simao et al.,2015),and Phylomarker(Lei et al.,2012).The resulting benchmark of single-copy genes will be instrumental for the assessment of genome assembly completeness and the study of fish systematics(Hughes et al.,2018).
In genome annotation,the variation of predicted gene and putative protein numbers should reflect the genomic nature in diverse fish species;however,it may be contributed by the prediction errors due to different algorithms used by various tools.A large portion of predicted genes or proteins were putative or functionally unknown.Functional validation of essential genes can be conducted using conventional approaches such as RT-qPCR or mutagenesis approaches such as CRISPRCas9(Cui et al.,2017).We propose to establish initiatives for functional annotation of fish genomes with a special interest in aquaculture or fisheries species.This community-based approach has been successfully demonstrated by the Zebrafish Information Network(ZFIN)project(Ruzicka et al.,2018)and the Functional Annotation of All Salmonid Genomes(FAASG)project(Macqueen et al.,2017).Such a communityengaged research initiative can avoid duplicated efforts in wholegenome annotation and functional gene validation.
There are dozens of important fisheries and aquaculture species with their draft genomes sequenced(Table S1);however,the genomes of many other species are not available or require improvements.These species include silver carp(Hypophthalmichthys molitrix),bighead carp(H.nobilis),Catla(Catla catla),and Roholabeo(Labeorohita),which are ranked the 2nd,5th,7th,and 10th in world aquaculture production,respectively(FAO,2018).The draft genomes of invasive silver carp and bighead carp were reported;however,the improvement of genome assemblies and additional sequencing of native fish are needed(Lu et al.,2020;Wang et al.,2019).The genomic data for Catla(Catla catla)and Roholabeo(Labeorohita)remain limited in public databases.
5.2.Genomics and aquaculture
Molecular markers play an essential role in the selection and breeding programs in aquaculture and have been broadly used to construct the linkage maps of important economic phenotypic traits such as growth,sex determination,and pathogen resistance(Yue,2014).The genetic gain was estimated to be greater than 12 percent per generation for growth rate and disease resistance through selective breeding in aquatic species(Gjedrem & Robinson,2014).Lu,Kuang,Zheng,Li,and Sun(2019)reviewed molecular marker-assisted breeding in aquatic species and proposed a practical approach in selective breeding.In a selective breeding experiment withC.carpio,two sequential selections in a pool of over 3 million individual mirror carp resulted in 300 phenotypically excellent individuals that were primarily from 15 families(Lu et al.,2019).The new stock grew more than 30% faster and exhibited superior genotypes enriched by more than 140% compared to the control group(Lu et al.,2019).The above genetic selection was conducted based upon only 20 molecular markers,which provides strong evidence of molecular marker-assisted selection being a powerful approach in selective breeding.
Genomic selection estimates individual breeding values using a large number of markers distributed across the genome(Meuwissen,Hayes,&Goddard,2001)and has a high accuracy of selection that can lead to rapid increased genetic gain(Sonesson,2011,pp.151-163).Genomic selection occurs at the population level and thus reduces generation intervals by the selection of progeny based on genotypes.Importantly,genome selection can predict the breeding potential of candidate populations based upon phenotypic data,which is particularly suitable for the selection of economic traits that are difficult to measure or count(Chen,Xu,&Liu,2019).When considering the cost of genomic selection versus traditional progeny testing,the associated costs could be reduced by as much as 90% of the original cost(Schaeffer,2006).However,its use in aquaculture species(yellow croak and Japanese flounder)has fallen behind compared to beef cattle and other livestock species(Rexroad,2019).Genome selection employs the prediction model based on genotypic and phenotypic data of a training population for the estimation of genomic estimated breeding values(GEBV)for all the individuals of the breeding population from their genomic profile(Bhat et al.,2016;Meuwissen et al.,2001).A suggested pipeline for genomic selection and breeding in aquaculture is shown in Fig.1,where the components related to different areas of science and technology were exemplified.
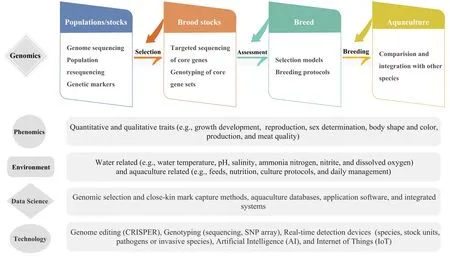
Fig.1.A proposed pipeline for fish selective breeding that involves genomics,phenomics,and other domains.Blue arrows indicate selective breeding processes,and orange arrows indicate feedback for further improvement of broodstock selection or breeding program assessment.The green trapezoid highlights genomic approaches and contributions towards improved selective breeding.The advances of aquaculture selection and breeding programs rely upon many other areas such as phenomes,environmental science,bioinformatics,statistics,and technologies.(For interpretation of the references to color in this figure legend,the reader is referred to the Web version of this article.)
One of the challenges in the application of genome selection is the reliability of phenotypic data used for training prediction models.Several phenotyping techniques have been developed in plants using non-invasive imaging,spectroscopy,image analysis,robotics,and high-computing facilities(Cobb,DeClerck,Greenberg,Clark,& McCouch,2013).These computer vision techniques have also been used in fish(Zenger,Khatkar,Jerry,& Raadsma,2017).Emerging technologies are needed for measuring the chemical(e.g.,fat,protein,moisture)and physical(e.g.,freshness,texture,color)attributes of fish with high accuracy(Saberioon,Gholizadeh,Cisar,Pautsina,& Urban,2017).We anticipate future computer vision intelligent systems will be able to extract quantitative information from digital images more accurately and thus can increase the accuracy of phenotypic data,which will lead to the improvement of genome selection(Fig.1).
Developing novel methods for the estimation of genomic breeding values(GEBV)has been quite active.A deep learning approach was developed to predict phenotypes from genotypes(Ma,Qiu,Song,Cheng,& Ma,2017).Comparative analysis of different algorithms has been conducted to predict genetic values in large yellow croaker(Dong,Xiao,Wang,& Wang,2016).Novel computing strategies were also developed for the prediction of GEBV,and the resulting computer program can be used by genomic selection programs(Dong,Fang,&Wang,2017).Genomic prediction models have been analyzed using multi-environment trials in chickpea that allowed the estimation of the effect of genotypic and environmental interaction,(Roorkiwal et al.,2018).The inclusion of genotypic and environmental effects in genome selection models should result in a more precise selection in aquaculture practice(Fig.1).
The whole-genome sequences offer a precious resource for molecular breeding through genome editing technologies.Genome editing technologies allow the interrogation of existing and novel genetic variation and thus facilitate the identification of causal genetic variation(Rexroad,2019).Genome editing has been used to identifydmrt1as an essential male sex-determining gene in Chinese tongue sole(Cui et al.,2017).Genome editing and transgenic technology are essentially different.Unlike the introduction of foreign genes in transgenic organisms,genome editing can mutate particular positions of a targeted gene and,therefore should be more easily accepted by consumers(Chen et al.,2019).The application of genome editing in aquaculture,although in its infancy,will undoubtedly become an essential means for the continued successful growth and stability of aquaculture production(Chen et al.,2019;Xu & Chen,2017;Yáñez,Newman,& Houston,2015).
5.3.Genomics and fisheries
World fisheries face the threats of overfishing and climate change that have resulted in the extinction of genetically unique stocks and loss of genetic diversity(FAO,forthcoming).Genomics has brought new tools that can help address fundamental questions in fisheries management such as stock identification,population structure,and adaptive response to environmental change(Bernatchez et al.,2017;Dudgeon et al.,2012;Li & Wang,2017).The identification of SNP markers through NGS has enhanced the ability to trace fisheries recourse or products to their original locations,allowing regulation enforcement in some commercially important fish species(Martinsohn,2011).Population genomic analysis through RAD-sequencing or genome resequencing has been applied in several important fisheries species including Asian seabass(Vij et al.,2016),European seabass(Tine et al.,2014),and Atlantic cod(Bradbury et al.,2010;Star et al.,2011)which unveiled the genetic basis of fisheries-induced evolution and the potential effects of environmental change.However,such an effort needs to be expanded to more species that are important in world fisheries.In particular,population genomic studies allow the detection of outlier loci and present a great promise for accurate genetic stock identification at a fine level of spatial resolution(Valenzuela-Quiñonez,2016).Fisheries genomic research shall focus on identifying informative SNPs that define management units and developing diagnostic markers for the monitoring of pathogens or invasive species(Bernatchez et al.,2017).
Besides the identification of species and the origin of stocks,fish abundance and spawning stock biomass are an important factor in fisheries management(Casey et al.,2016).The use of genetic markers to identify close-kin relationships provides fishery-independent estimates of spawning stock biomass(Ovenden,Berry,Welch,Buckworth,& Dichmont,2015).The close-kin approach has been applied to estimate the spawning stock biomass of Southern Bluefin tuna(Bravington,Grewe,&Davies,2013;Bravington,Grewe,&Davies,2016).The closekin method estimates the stock abundance based upon the genotypic data of sampled individuals,where the genotype of an individual can be considered a capture of the genotypes of each of its parents(Skaug,2001).However,this method requires a large number of samples in order to find sufficient numbers of parent-offspring pairs.Further evaluation of the close-kin method,including its use in the estimation of fish abundance,is needed in the future(Casey et al.,2016).
Applying genomic approaches to fisheries management is feasible and cost-efficient in most cases;however,the transformation of genomic findings into management practices has stagnated(Bernatchez et al.,2017).The genomic tools and their power in species identification,determination of management units,and evaluation of natural resources shall give full consideration when fisheries management policies and guidelines are made(Martinsohn,2011).The stakeholders including managers and fisheries geneticists need to work collaboratively and make sure genomic tools become an integral component of fisheries management in the future.
5.4.Integrated and intelligent system for aquaculture and fisheries
Rapid advancements of next-generation sequencing technologies and broad interests in sequencing genomes of economically,ecologically,or evolutionarily significant species have made available hundreds of fish draft genomes in public repositories.The majority of fish genomes can be found in the genome database at the U.S.National Center for Biotechnology Information(NCBI),which has published the genomic data of 265 fish species,including 64 chromosome-level genome sequences(as of Dec 17th,2019;Table S2).Another important genome repository is Ensembl,which has made available approximately 60 fish genomes to the public(Table S1).However,the genome databases in NCBI and Ensembl are developed to serve diverse research communities and may not fulfill the needs of the aquaculture and fisheries community.In this respect,a few species-specific genome resources such as GCGB(Grass Carp Genome Database)(Chen et al.,2017)and SalmoBase(a molecular data resource for salmonid species)(Samy et al.,2017)have been developed,which allows accessing genomic data,linkage mapping,and gene expression data.A database system dedicated to fish that integrates genetic,phenotypic,and environmental data is currently lacking.It is thus important to develop an integrated big data platform for major species in aquaculture and fisheries(Chen et al.,2019).Ag-BioData is such a database system in agriculture,which could be adopted to aquaculture and fisheries that will enhance genomics,genetics,and breeding research outcomes through standardization of protocols and practices(Harper et al.,2018).Moving forward with emerging technologies of data science and artificial intelligence(AI),the fisheries and aquaculture communities should strengthen collaborations and develop cloud sourcing projects to tackle challenging issues such as data sharing,integration and use in fisheries and aquaculture and promote technological innovations such as IoT(Internet of Things)for Aquaculture 4.0(Fig.1;Dupont,Cousin,& Dupont,2018).
6.Conclusions
The advances of next-generation sequencing technologies and genomics have revolutionized fisheries and aquaculture sciences and practices.We now have several dozens of important aquaculture and fisheries species with the complete genomes sequenced and available for analysis,comparison,and knowledge discovery.We have gained much knowledge about genomic mechanisms involved in germplasm resource utilization,disease resistance,growth and development,sexual determination,and fisheries management.The full potential of genomic applications in genomic selection and fisheries management,however,has not been achieved.In the coming decades,the application of genomic techniques such as genome editing and genomic selection,along with the use of emerging big data and artificial intelligence systems,are expected to leverage considerably sustainable breeding programs and achieve the goal of eradicating global poverty by 2030.
Acknowledgments
We are grateful to Drs.T.D.Kocher,Z.Liu,K.S.Jakobsen,and Y.Nakamura who kindly provided us updates about their fish genome projects and the latest references.We also want to thank Mary Awsiukiewicz for proofreading the manuscript.This publication was made possible through funding support from the National Science Foundation(DBI-1919574)and the University of Nebraska at Omaha.GL wants to thank Ms.Y.Liu in the Editorial Office ofAquaculture and Fisheriesfor the invitation to contribute this review.
Appendix A.Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.aaf.2020.05.004.
 Aquaculture and Fisheries2020年4期
Aquaculture and Fisheries2020年4期
- Aquaculture and Fisheries的其它文章
- The economic contribution of fish and fish trade in Bangladesh
- Eye location,cranial asymmetry,and swimming behavior of different variants of Solea senegalensis
- Effect of chicken haemoglobin powder on growth,feed utilization,immunity and haematological index of largemouth bass(Micropterus salmoides)
- Effect of the number of freeze-thaw cycles number on the quality of Pacific white shrimp(Litopenaeus vannamei):An emphasis on moisture migration and microstructure by LF-NMR and SEM
- Hydrodynamic performance and structural response characteristics of the double-slotted vertical cambered V-Type otter board