
早期使用不同疗程抗生素对早产儿日龄28d肠道菌群的影响
2020-08-04 13:55王俊平王艳丽钟隽镌
中国当代医药 2020年17期
王俊平 王艳丽 钟隽镌
[摘要]目的 利用16S rRNA測序技术,探讨早期经验性应用抗生素不同疗程对早产儿日龄28 d肠道微生物的影响。方法 选取2017年12月~2018年5月在广东省妇幼保健院新生儿科住院治疗的27例早产儿作为研究对象,其中使用青霉素+三代头孢菌素≤3 d为短疗程组(G1组,9例),使用青霉素+三代头孢菌素≥7 d为长疗程组(G2组,18例)。收集28 d日龄的粪便标本,采用16S rRNA测序技术分析菌群组成的变化。结果 菌群的结构组成中,主要的菌门包括放线菌门、拟杆菌门、厚壁菌门、变形菌门,其中厚壁菌门含量最为丰富。G1组中的拟杆菌门(0.03%)和变形菌门(34.11%)与G2组(0.04%、14.23%)比较,差异无统计学意义(P>0.05)。G1组和G2组的alpha多样性比较,差异无统计学意义(P>0.05)。G1组中的β-变形菌纲(LDA=6.43,P=0.017)、丛毛单菌科(LDA=6.24,P=0.017)、皮杆菌科(LDA=6.60,P=0.016)、黄体杆菌属(LDA=6.06,P=0.026)和韦荣球菌属(LDA=7.51,P=0.049)高于G2组,差异有统计学意义(P<0.05)。结论 早期经验性、长期应用广谱抗生素可能会导致早产儿肠道正常菌群的定植和达优势化时间延迟。
[关键词]抗生素;肠道菌群;早产;16S rRNA测序
[中图分类号] R722.6 [文献标识码] A [文章编号] 1674-4721(2020)6(b)-0008-04
[Abstract] Objective To explore the influence of early empirical application of different treatment courses of antibiotic on the gut microbiota of 28-day-old preterm infants through 16S rRNA sequencing technology. Methods From December 2017 to May 2018, a total of 27 cases of hospitalized preterm infants in Guangdong Women and Children Hospital were enrolled, among them, penicillin + the third generation cephalosporin ≤ 3 days were selected as the short course group (the G1 group, 9 cases), penicillin + the third generation cephalosporin ≥ 7 days were selected as the long course group (the G2 group, 18 cases). The fecal samples at postnatal 28 days were collected, and 16S rRNA sequencing technology was used to analyze the changes of microbial community diversity. Results In the structural composition of the flora, the main phylum included Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria, among which pachytene was the most abundant, the thick-walled mycoplasma was the most abundant. There were no significant differences in the Bacteroidetes and Proteobacteria between the G1 group (0.03%, 34.11%) and the G2 group(0.04%, 14.23%) (P>0.05). There was no statistical difference in the alpha diversity between the G1 group and the G2 group (P>0.05). Betaproteobacteria (LDA=6.43, P=0.017), Comomamonadaceae (LDA=6.24, P=0.017), Dermabacteraceae (LDA=6.60, P=0.016), Luteibacter (LDA=6.06, P=0.026) and Veillonella (LDA=7.51, P=0.049) in the G1 group were higher than those in the G2 group, there were statistical differences (P<0.05). Conclusion Early empirical and long-term application of broad-spectrum antibiotic may lead to the colonization of normal intestinal flora and the delay of the advantage of time in preterm infants.
[Key words] Preterm infant; Gut microbiota; Antibiotic; 16S rRNA sequencing
宫内感染是早产的一个常见原因,脓毒症是早产儿死亡和发病的主要原因之一,许多早产儿出生就有感染的嫌疑。经验性抗生素治疗是新生儿病房常见的预防和治疗脓毒症的方法[1],但过多的抗生素治疗对早产儿肠道菌群发育不利,可能对长期健康造成不良影响。已有研究发现,每使用抗生素治疗1 d就会使晚发性败血症(LOS)、新生儿坏死性小肠结肠炎(NEC)或死亡风险增加1.24倍[2]。目前,抗生素对早产儿肠道菌群影响的研究主要集中在对早产儿出生14 d内的影响,而早产儿晚发性败血症和新生儿坏死性小肠结肠炎更常发生于14 d之后,因此,了解抗生素对早产儿14 d之后肠道菌群的影响至关重要。本研究利用16S rRNA测序技术,旨在探讨早期经验性使用抗生素不同治疗时间对早产儿出生后28 d肠道菌群结构组成的影响。
1资料与方法
1.1一般资料
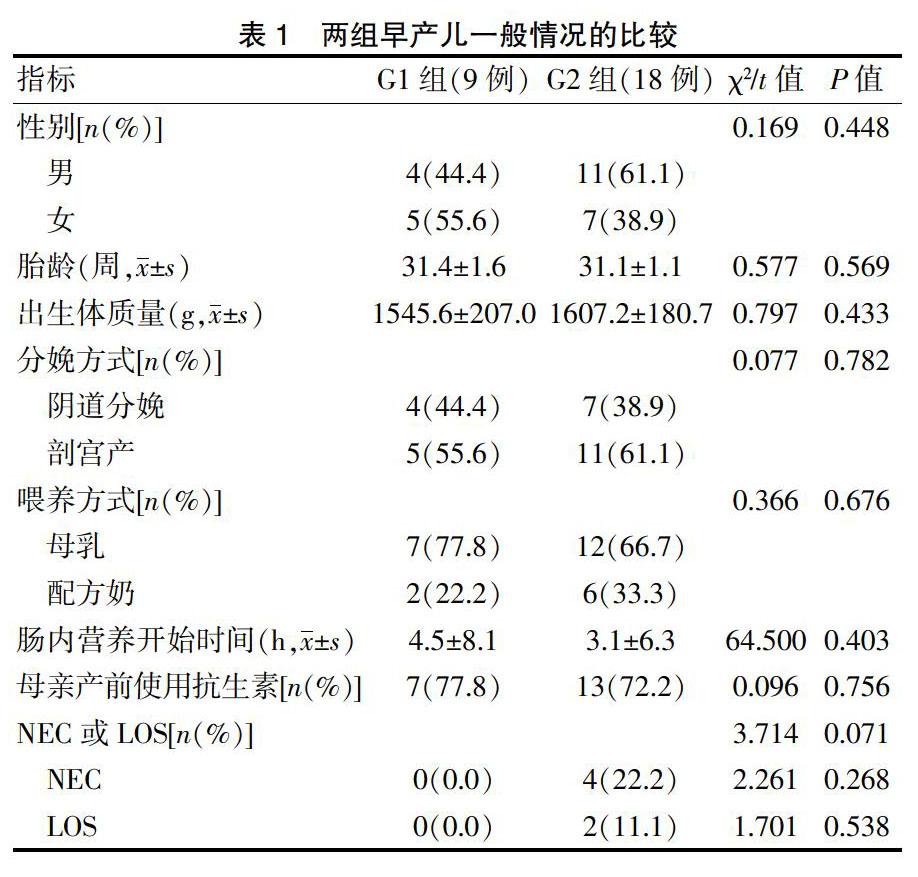
选取2017年12月~2018年5月在广东省妇幼保健院出生并住院治疗的早产儿作为研究对象,研究期间符合入选标准的早产儿共34例,未达研究终点签字出院或因其他原因死亡者7例,最终纳入27例。将出生后24 h内使用青霉素+三代头孢菌素≤3 d的早产儿作为短疗程组(G1组,9例),将出生后24 h内使用青霉素+三代头孢菌素≥7 d的早产儿作为长疗程组(G2组,18例)。两组受试者的性别、胎龄、出生体质量、分娩方式、母亲产前抗生素使用、开奶时间、NEC、LOS等一般资料比较,差异均无统计学意义(P>0.05)(表1)。本研究获广东省妇幼保健院伦理委员会批准,且获得患儿父母或法定监护人的书面知情同意。早产儿经验性应用抗生素的指征:存在感染危险因素(母亲分娩时体温≥38℃,胎膜早破>18 h,绒毛膜羊膜炎等)和临床表现怀疑早期感染。新生儿败血症诊断标准:依据《新生儿败血症诊断及治疗专家共识》(2019年版)制订[1]。G1组纳入标准:①在我院出生的早产儿;②有经验性抗生素治疗指征;③出生48 h内白细胞计数、血小板、C反应蛋白(CRP)、降钙素原连续检测2次,<2项阳性,血培养阴性;④出生24 h内使用青霉素+三代头孢菌素≤3 d。G2组纳入标准:①在我院出生的早产儿;②有经验性应用抗生素治疗指征;③出生48 h内白细胞计数、血小板、CRP、降钙素原检测,其中至少2项阳性;④出生24 h内使用青霉素+三代头孢菌素,且应用≥7 d。两组排除标准:①临床需要使用微生态制剂;②存在先天畸形、先天异常或遗传代谢性疾病者。
1.2方法
1.2.1样本提取、DNA提取、PCR和测序 使用无菌标本瓶采集受试者出生后第28天的粪便样本,量不少于5 g,未排便者使用开塞露加生理盐水通便。采集后立即冰冻于-20℃,并尽快冻存于-80℃冰箱。本实验根据Magen生物公司的HiPure Stool DNA Kit试剂盒说明提取粪便细菌总DNA。提取好DNA的量用Nano Drop ND-1000分光光度计测定浓度,共分析粪便样本27份。对浓度符合要求的样品进行PCR扩增,主要针对性扩增16S的V3~V4可变区,选用的扩增引物是338F(5′-ACTCCTACGGGAGGCAGCAG-3′)和806R(5′-GGACTACHVGGGTWTCTAAT-3′)。PCR体系是50 μl,含有25 μl 2× Premix Taq,每种引物2 μl,DNA模板3 μl。PCR产物的完整性、片段长度和浓度用1.0%的甘蔗凝胶电泳进行检查。浓度和条带长度符合预期范围的样品等浓度混合,然后上机Illumina Hiseq 2500平台采用2×250的模式测序。
1.2.2数据分析 首先将利用Trimmomatic软件[3]测序得到的Raw Reads进行质量过滤,过滤掉含N、质量值低于20及过滤后序列长度低于100 bp的reads。质控后的clean reads利用FLASH软件[4]基于overlap关系进行拼接。基于overlap区允许最大错配率为0.1,利用软件Mothur[5]对序列进行样品对分配,同时控制barcode的最大错配数为2,最大引物错配数为3进行筛选。随后利用软件usearch[6]将筛选得到的tags进行聚类,97%及以上的一致性序列聚成一个OTU,在usearch聚类的同时,去除含嵌合体的tags。最后利用软件QIIME[7]中的assign_taxonomy.py基于Silva数据库对OTU进行物种注释,并去掉注释为叶绿体或线粒体及不能注释的OTU,最终得到序列注释表。样品的alpha多样性指数用QIIME的alpha_diversity.py完成,beta diversity的距离利用QIIME中的beta_diversity.py计算得到。软件LEfSe[7]用于进行差异菌群的分析,阈值设定LDA≥2。
1.3观察指标
采用16S rRNA测序技术对出生后28 d龄粪便标本进行检测,考察出生后经验性应用不同抗生素及不同治療时间对早产儿肠道菌群的影响,主要观察指标包括菌群的结构组成、菌落的多样性及差异菌。
1.4统计学方法
采用SPSS 16.0统计学软件进行数据分析,符合正态分布的计量资料用均数±标准差(x±s)表示,两组间比较采用t检验;不符合正态分布的计量资料采用中位数表示,两组间比较采用Mann-Whitney U检验;计数资料采用率表示,组间比较采用Fisher精确检验,以P<0.05为差异有统计学意义。
2结果
2.1两组菌群结构组成的分析
在所有研究样本中,主要的菌门包括放线菌门(Actinobacteria)、拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)、变形菌门(Proteobacteria),详见图1A(封三)。其中厚壁菌门含量最为丰富,其在G1组的中位数值为65.54%,基本与G2组无区别(66.60%,P=0.18)。同时拟杆菌门在两组中也基本无变化(0.03% vs.0.04%,P=0.96)。放线菌门在G1组的中位数值为0.16%,略低于G2组的0.25%(P=0.14)。此外,变形菌门(34.11% vs.14.23%,P=0.25)含量在G2中比G1组亦有所增加。主要菌属包括拟杆菌属(Bacteroides)、艰梭菌属(Clostridioides)、梭状芽孢杆菌属(Clostridium_sensu_stricto_1)、肠杆菌属(Enterobacter)、肠球菌属(Enterococcus)、埃希菌属(Escherichia)、克雷伯菌属(Klebsiella)、Robinsoniella属、链球菌属(Streptococcus)和韦荣球菌属(Veillonella),详情见图1B(封三)。相比于G1组,G2组中的拟杆菌属(0.016% vs. 0.029%,P=0.46)、梭状芽孢杆菌属(0.97% vs. 1.06%,P=0.84)、埃希菌属(0.27% vs. 1.38%,P=0.09)、克雷伯菌属(0.32% vs. 2.06%,P=0.67)和艰梭菌属(0.36% vs. 0.47%,P=0.70)含量的中位数值含量有所下降。与之相反的是,链球菌属的中位数值(1.00% vs. 0.41%,P=0.59),肠球菌属(2.06% vs. 1.34%,P=0.33)和Robinsoniella属(0.90% vs. 0.30%,P=0.09)含量的中位数值在G2组中相对较高。
2.2两组菌落多样性的比较
alpha多样性综合菌群的种类和含量的均一性常用Shannon和Simpson两个指数来表征。G1组中的Shannon指数平均值为2.43±1.03,与G2组的2.41±1.04比较,差异无统计学意义(P=0.96)(图2A,封三)。同时,G1组中Simpson指数的平均值为0.38±0.20,与G2组的0.61±0.22比较,差异无统计学意义(P=0.79)(图2A,封三)。为了分析菌群整体的差异,本研究进行了PCoA分析,结果见图2B(封三),图中显示G1组与G2组的样品重叠在一起,未能区分开,显示两组样品的菌群结构较为接近。
2.3两组差异菌的比较
为了近一步分析G1组与G2组的菌群差异,本研究利用LEfSe软件进行分析,发现G1组中的β-变形菌纲(Betaproteobacteria,LDA=6.43,P=0.017)、丛毛单菌科(Comamonadaceae,LDA=6.24,P=0.017)、皮杆菌科(Dermabacteraceae,LDA=6.60,P=0.016)、黄体杆菌属(Luteibacter,LDA=6.06,P=0.026)和韦荣球菌属(Veillonella,LDA=7.51,P=0.049)显著高于G2组(图3,封四)。
3讨论
早产儿肠道功能不完善,肠道菌群定植明显延迟,使得其微生物群更容易受到破坏[8]。由于早产儿免疫功能不完善、对感染抵抗力差,且早发败血症诊断困难,常需要出生后即开始经验性应用第三代头孢菌素等广谱抗生素。因第三代头孢菌素抗菌谱广,可能导致早产儿肠道菌群定植的模式及数量发生异常改变,从而增加早产儿新生儿坏死性小肠结肠炎、诱导耐药产生、继发真菌感染等严重并发症的发生率,导致早产儿病死率增高。已有研究显示,早产儿肠道菌群的建立及细菌的异常定植与LOS和NEC有关[9-10]。本研究结果显示,G2组有4例NEC,2例LOS,G1组无NEC和LOS病例。
早产儿微生物群的多样性比足月新生儿更有限,早产儿肠道正常菌群的定植明显延迟,达优势化时间也延迟;与足月新生儿相比,差异包括厌氧菌减少、厚壁菌和变形菌的丰度增加、拟杆菌的丰度减少以及双歧杆菌定植时间的显著延迟[11-12]。有研究显示,在使用抗生素和不使用抗生素的对照婴儿之间的alpha多样性差异有统计学意义[13]。本研究结果显示,在日龄28 d时,两组肠道菌群的alpha多样性差异无统计学意义,考虑与本研究样本量少、早产儿胎龄较低及均使用抗生素有关。
在肠道菌群结构组成方面,本研究结果显示两组早产儿的肠道菌群均以厚壁菌门和变形菌门为主,拟杆菌门和放线菌门的丰度较低,且本研究未检测到显著的双歧杆菌;G2组的专性厌氧菌中的拟杆菌属、梭状芽孢杆菌属含量降低,埃希菌属、克雷伯菌属相对丰度低;而肠球菌属在G2组的比例高于G1组,这与唐小丽等[14]和朱丹萍等[15]的研究结果一致。肠球菌细胞壁坚厚,对许多抗菌药物大多表现为固有耐药,抗生素可将肠道内共生菌杀灭,从而导致了上述耐藥细菌及致病菌的过度生长,说明使用抗生素尤其是长期使用后早产儿肠道定植更多的兼性厌氧菌,严格厌氧菌定植延迟。两组差异菌比较显示,G1组中的β-变形菌纲、丛毛单菌科、皮杆菌科、黄体杆菌属和韦荣球菌属显著高于G2组。韦荣球菌属是乳酸发酵菌,先前的一些研究已报道了其是成人和婴儿重要的肠道定植菌[16],提示长期使用广谱抗生素导致早产儿肠道正常菌群的定植明显延迟,达优势化时间也明显延迟。
本研究留取早产儿出生后28 d的粪便样本,结果表明早期长时间广谱抗生素尤其是第三代头孢菌素对早产儿肠道菌群的影响至少可持续到出生后28 d,其主要的影响以双歧杆菌、乳酸菌、革兰阴性杆菌为主。与长期使用抗生素对早产儿微生物组成的影响持续了整个出生后的前6周[17]结果一致。
综上所述,经验性、长期应用广谱抗生素可能会导致早产儿肠道菌群定植的模式及数量发生较长期的异常改变,应谨慎对待早产儿的广谱抗生素治疗,并且必须尽一切努力将治疗时间限制在最短。由于本研究分析的病例数较少,均为使用抗生素的病例,且无动态观察抗生素对早产儿肠道菌群的影响,还需要更多的病例及样本来验证这些结果。
[参考文献]
[1]中华医学会儿科分会新生儿学组,中国医师协会新生儿科医师分会感染专业委员会.新生儿败血症诊断及治疗专家共识(2019版)[J].中华儿科杂志,2019,57(4):252-257.
[2]Joseph B,Cantey MD,Alaina K,et al.Early antibiotic exposure and adverse outcomes in preterm very low birth weight infants[J].J Pediatrics,2018,203(12):62-67.
[3]Bolger AM,Lohse M,Usadel B.Trimmomatic:a flexible trimmer for lllumina sequence data[J].Bioinformatics,2014,30(15):2114-2120.
[4]Magoc T,Saizberg SL.FLASH:fast length adjustment of short reads to improve genome assemblies[J].Bioinformatics,2011,27(21):2957-2963.
[5]Schloss PD,Westcott SL,Ryabin T,et al.Introducing mother:open-source,platform-independent,community-supported software for describing and comparing microbial communities[J].Appl Environ Microbiol,2009,75(23):7537-7541.
[6]Edgar RC.Search and clustering orders of magnitude faster than BLAST[J].Bioinformatics,2010,26(19):2460-2461.
[7]Caporaso JG,Kuczynski J,Stombaugh J,et al.QIIME allows analysis of high-throughput community sequencing data[J].Nat Methods,2010,7(5):335-336.
[8]Chernikova DA,Madan JC,Housman ML,et al.The premature infant gut microbiome during the first 6 weeks of life differs based on gestational maturity at birth[J].Pediatr Res,2018,84(1):71-79.
[9]Berrington JE,Hearn RI,Bythell M,et al.Deaths in preterm infants:changing pathology over 2 decades[J].J pediatr,2012,160(1):49-53.
[10]Grishin A,Papillon S,Bell B,et al.The role of the intestinal microbiota in the pathogenesis of necrotizing enterocolitis[J].Semin pediatr Surg,2013,22(2):69-75.
[11]Groer MW,Luciano AA,Dishaw LJ,et al.Development of the preterm infant gut microbiome:a research priority[J].Microbiome,2014,2:38.
[12]Claud EC,Keegan KP,Brulc JM,et al.Bacterial community structure and functional contributions to emergence of health or necrotizing enterocolitis in preterm infants[J].Microbiome,2013,1(1):20.
[13]Mohan P,Julia C,Phillip IT,et al.Intestinal dysbiosis in preterm infants preceding necrotizing enterocolitis:a systematic review and meta-analysis[J].Microbiome,2017,5(1):31.
[14]唐小麗,余加林,艾青,等.肠道菌群多样性在喂养不耐受新生儿中的作用[J].第三军医大学学报,2014,36(20):2133-2137.
[15]朱丹萍,杜立中,余加林,等.早产儿早期经验性应用抗生素对其肠道菌群的近期影响[J].中国循证儿科杂志,2016,11(1):26-29.
[16]Rinttila T,Kassinen A,Malinen E,et al.Development of an extensive set of 16S rDNA-targeted primers for quantification of pathogenic and indigenous bacteria in faecal samples by real-time PCR[J].J Appl Microbiol,2004,97(6):1166-1177.
[17]Zwittink RD,Renes IB,van Lingen RA,et al.Association between duration of intravenous antibiotic administration and early-life microbiota development in late-preterm infants[J].Eur J Clin Microbiol Infect Dis,2018,37(3):475-483.
(收稿日期:2020-03-09 本文编辑:祁海文)
猜你喜欢
中国典型病例大全(2022年10期)2022-05-10
医学食疗与健康(2022年2期)2022-04-23
中国药学药品知识仓库(2022年5期)2022-04-11
大众健康(2021年8期)2021-08-04
中国保健营养(2019年7期)2019-10-21
大众医学(2019年3期)2019-03-29
大众健康(2018年8期)2018-08-03
百科知识(2017年10期)2017-05-19
百科知识(2017年3期)2017-03-17
家庭用药(2016年8期)2016-05-14
