
儿童肺动脉高压定义和诊断及治疗的研究进展
2020-07-28 06:35庹虎唐其柱何兵杨青黎文静
实用心脑肺血管病杂志 2020年7期
庹虎,唐其柱,何兵,杨青,黎文静
肺动脉高压(PAH)是一类以肺动脉压力(PAP)、肺血管阻力(PVR)进行性升高为主要特点的肺血管疾病,其是肺血管疾病常见的病理生理状态,可导致患者右心功能衰竭甚至死亡[1]。PAH在儿童中起病隐匿、病因复杂、发病率高且预后不佳而备受关注。近年来,由于心导管检查的普及和基因诊断技术的开展,PAH的诊断越来越精准化。同时随着靶向治疗的出现,PAH的治疗学得到了较快发展。本文主要综述了儿童PAH的定义、诊断及治疗现状,以期为儿童PAH规范化诊疗提供借鉴。
1 儿童PAH定义及分类
1.1 儿童PAH定义 目前普遍采用的是2008年在美国第4届全球PH会议上制定的PAH诊断标准,即在海平面、静息状态下,肺动脉平均压(mPAP)≥25 mm Hg(1 mm Hg=0.133 kPa),肺毛细血管楔压(PCWP)正常(≤15 mm Hg),PVR升高[2]。胎儿mPAP与PVR均处于较高水平,出生后随着血液重新分布,PAP逐步下降,出生后2~6个月PAP水平可降至参考范围内。受多种因素影响(如先天性心脏病、呼吸窘迫综合征、支气管肺发育不良、胎粪吸入性肺炎等)胎儿血液循环状态向成年人过渡时常发生障碍,使PAP不能降至参考范围内,同时因为儿童体循环压力明显低于成年人,所以在相同mPAP下,单纯根据mPAP来定义儿童PAH是不合理的[3]。2011年Panama肺血管研讨会指出,在mPAP<25 mm Hg时,若肺血管阻力指数(PVRI)≥3 wood单位或跨肺压>6 mm Hg可诊断为儿童PAH[4]。
1.2 PAH的分类 1973年世界卫生组织(WHO)在日内瓦召开的首届PAH世界论坛中制定了PAH的分类标准,主要为原发性PAH和继发性PAH[3]。1998年WHO在法国的依云第二次PAH专家会议上对PAH的分类做出了修改,主要为:第1类为原发性PAH,包括原因不明的PAH,可分为散发型和家族型;第2类是与多种基础疾病相关的PAH,包括结缔组织病、先天性体-肺分流、门静脉高压症、人类免疫缺陷病毒(HIV)感染相关性PAH、药物和毒素性PAH、新生儿持续性PAH等[5]。而2003年WHO在威尼斯举办的第三次PAH专家工作组会议中又提出了新的修订,最大特点是将原发性PAH更改为特发性肺动脉高压(idiopathic pulmonary hypertension,IPAH)[6]。随着对疾病病理生理和诊断技术的研究发展,PAH标准并不能满足临床需求,2009年8月欧洲心脏病学会(ESC)年会发布了新的PAH诊断指南,其根据PAH的病理、临床表现和治疗措施将PAH分为5大类,即动脉性PAH、左心疾病所致PAH、肺部疾病或低氧血症所致PAH、慢性血栓栓塞性PAH、原因不明和/或多种机制所致的PAH[7],但此项分类是否符合儿童PAH的发病特点还需要进一步商榷。
2 儿童PAH的诊断
PAH早期症状多不典型,发病通常较隐匿,绝大多数患儿在疾病的早期阶段仅表现为气促、发绀、心动过速等,并无明显特异性或相关病理生理指标改变,当患儿出现明显临床症状(如乏力、活动耐力下降、晕厥甚至下肢水肿、腹腔积液)时,其心功能已达到WHO分级的Ⅲ~Ⅳ级,因此,寻找疾病诊断生物学标志物对提高儿童PAH的诊断率具有重要意义。
PAH常用的诊断方法有胸部X线、心电图、多普勒超声心动图、心脏磁共振成像(MRI)及右心导管检查。胸部X线和心电图检查常缺乏特异性,而右心导管检查在基层医院难以实施,因此心脏多普勒超声心动图检查是诊断PAH应用最广、操作最简便的无创性检查。目前心脏多普勒超声心动图检查有3种方法:(1)心脏和大血管间压力阶差测定法,此法依赖于右房室瓣反流或心内分流,有一定局限性;(2)右心室收缩时间间期估测法;(3)右心室等容舒张时间测定法,由于影响心脏舒张时间的因素较多,其临床应用较少[8]。近年来MRI在心血管疾病诊断中的地位越来越突出,其可以清晰地显示心脏各腔空间状况、血流走形、血管的解剖结构,常用来作为PAH患者的随访手段。右心导管检查是诊断PAH的“金标准”[9],其可以直接检测血管管腔内压力及血流动力学数据,同时对PAH的病因分析、疾病分度、治疗效果的监测均具有明确的指导作用。
随着分子医学的发展及基因测序的广泛应用,基因诊治技术作为一种新型手段在PAH诊断中的价值逐渐显现。目前已证实微小核糖核酸(miRNAs)、骨形成蛋白Ⅱ型受体基因(BMPR2 gene)、激活素受体样激酶1基因(ACVRL1/ALK1)及 ENG、KCNA5、KCNK3、SMAD4、SMAD9、CAV1、GDF2、NOTCH3、EIF2AK4等基因与PAH发病有不同程度的相关性[10-14]。
3 儿童PAH的治疗
PAH的初始治疗是针对基础疾病和诱发的因素进行治疗,对于低氧血症尤其是阻塞性睡眠障碍患儿给予吸氧;对于慢性复发性血栓栓塞患儿和IPAH患儿给予抗凝治疗;对于感染引起的PAH患儿积极控制感染;对于先天性心脏病患儿可通过介入治疗或手术纠正血流动力学改变。而内科治疗PAH的手段主要有两个方面:(1)靶向肺血管舒张药物,此类药物选择性作用于异常表达的内皮素通路、一氧化碳/环磷酸鸟苷通路(NO/cGMP)、前列环素通路,使肺血管舒张,保证正常肺的血液供应;(2)靶向抗肺血管重塑药物,针对酪氨酸激酶等受体抑制肺血管增生、肥厚,减缓血管狭窄的发生从而减小PAP。2007年前,全球上市治疗PAH的药物只有静脉注射依前列醇、皮下注射和静脉注射曲前列尼尔、吸入伊洛前列素,而随着病理生理和分子机制的研究,新的药物陆续上市,PAH的治疗也进入多元化阶段[15]。
目前已上市的治疗PAH的药物包括钙离子通道阻滞剂(CCB)、前列环素(PGI2)及其类似物、内皮素受体拮抗剂(ERA)、5型磷酸二酯酶(PDE5)抑制剂以及一氧化氮(NO)[16]。CCB自20世纪80年代便应用于IPAH的治疗,而CCB可使慢性阻塞性肺疾病患儿的通气/血流比失调,肺部气体交换发生障碍,因此使用前需要完善血管扩张试验,阳性者则可谨慎用药,且对儿童治疗的有效性也非常有限。NO的t1/2较短[13],需要吸入的时间较长,操作方法复杂,价格昂贵,在有条件的治疗室内可以采用此法,但不适合一般患儿的长期使用。因此临床常将PGI2及其类似物、ERA、PDE5抑制剂作为靶向药物来治疗儿童PAH,具体治疗方法见表1[17-25]。
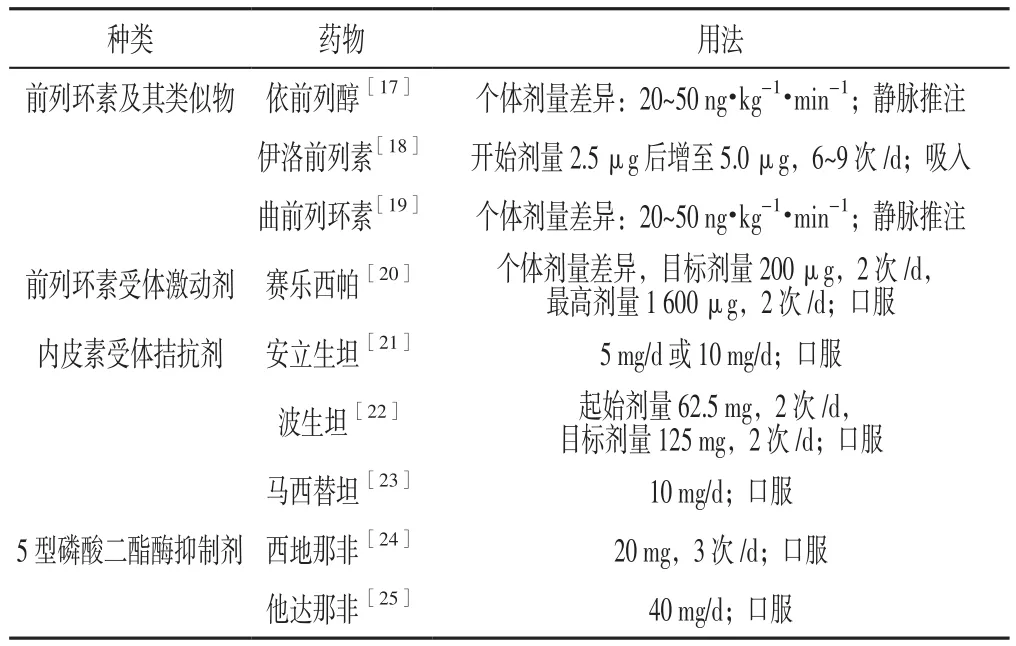
表1 前列环素及其类似物、前列环素受体激动剂、内皮素受体拮抗剂、5型磷酸二酯酶抑制剂治疗儿童PAH的方法Table 1 Treatment of PAH in children with prostacyclin and its analogues,prostacyclin receptor agonist,endothelin receptor antagonists,and phosphodiesterase type 5 inhibitors
3.1 PGI2及其类似物 PGI2及其类似物在PAH治疗上具有里程碑式的作用,其是一种花生四烯酸代谢物,是强效的内皮源性血管扩张因子,可通过环磷酸腺苷途径活化蛋白激酶A,进而致平滑肌舒张并抑制平滑肌细胞增殖,此外,PGI2能抗血小板聚集以预防血栓形成[26]。依前列醇(prostaglandin)是第一个被证实治疗PAH有效的药物,也是第一个被美国食物药品监督管理局(FDA)批准用于治疗PAH的PGI2类似物,其提高了重度心功能不全患者的存活率,且3年存活率达63%[15]。根据患儿的耐受情况一般从1~2 ng·kg-1·min-1开始,最高可达 5~10 ng·kg-1·min-1,PGI2 的 t1/2较短(3~5 min),需长期静脉注射维持,而建立中心静脉通道增加了感染、血栓形成等并发症的发生率。且临床观察发现,突然停药后可导致患儿PAP的复发[27],同时在治疗过程中药物需低温储存,因此限制了依前列醇在临床的大量应用。美国哥伦比亚大学的一项针对儿童静脉使用依前列醇的长期随访研究表明,PAP患儿4年生存率为94%,10年生存率为37%。贝前列素(beraprost)是可以口服的PGI2类似物,一般应用于症状较轻的患儿,主要不良反应包括头晕、腹泻、心悸及面部潮红等[28],长期应用的疗效及安全性缺少大样本的临床研究。曲前列素(treprostinil)是PGI2的一种剂型,在室温下稳定,且t1/2达4 h,可以通过皮下注射、静脉注射及吸入给药。CHANNICK等[29]的一项研究表明,健康志愿者吸入的曲前列素的生物利用率为64%~72%,长期吸入曲前列素可改善临床症状和血流动力学。国外一项包括470名PAH患儿的双盲随机对照研究表明,用药12周后,曲前列素治疗组患儿的6 min步行距离长于安慰剂组[24]。如果使用其他类型靶向药物效果不佳时可以考虑加入吸入性曲前列素,而且社区门诊也可以配备雾化吸入设备,其临床应用前景更加广泛[30]。伊洛前列素(iloprost)是PGI2类似物应用最多的吸入性制剂,因其t1/2短,每天需吸入6~12次,对患者配合度要求较高,这对儿童较为困难。美国FDA已经通过认证,欧洲也批准其用于Ⅲ级IPAH患儿,且目前在我国也已经上市[31]。
3.2 ERA PAH患儿的肺动脉内皮细胞内皮素1(ET-1)水平和血浆ET-1水平均有不同程度升高,而内皮素(ET)不仅可引起肺血管的收缩,还可促进和导致肺血管重构,阻断ET受体是治疗PAH的另一种重要方法。ET有两个受体,即内皮素受体A(ETR-A)和内皮素受体B (ETR-B),前者可导致血管持续收缩和平滑肌细胞增殖;后者被认为主要参与PAH血管内瘢痕形成,同时介导细胞产生NO和PGI2而使血管扩张。目前波生坦(bosentan)在临床中应用最为广泛,作为一种非选择性ERA,其同时作用于ETR-A和ETR-B,儿童常用剂量为:体质量<10 kg时给予15.60 mg,2次 /d;10~20 kg时给予 31.25 mg,2次 /d;>20~40 kg时给予62.50 mg,2次/d;>40 kg时给予125.00 mg,2次/d。波生坦的主要不良反应为肝功能损伤,因此用药期间需要定期监测肝功能情况[32]。近年来选择性ERA也逐步应用于临床,包括安贝生坦(ambrisentan)和西他生坦(sitaxsentan),不良反应较波生坦小,且目前使用的国产安贝生坦给患儿及家属带来的经济压力也逐步降低。
3.3 PDE-5抑制剂 西地那非(sildenafil)属于PDE-5抑制剂,主要通过增强NO/NO/cGMP来扩张肺血管,从而改善肺循环。2005年,美国FDA批准西地那非用于PAH治疗,并作为WHO PAH功能分级Ⅱ~Ⅲ级患者的一线治疗用药,儿童剂量为0.25~1.00 mg/kg,3次/d[33]。国内外相关临床研究表明,西地那非可不同程度改善PAH患者血流动力学及运动耐量[34],王银谦等[35]通过收集2000—2012年西地那非治疗PAH患儿的文献进行Meta分析,结果表明,患儿用药后的mPAP、6 min步行距离及活动耐量明显提高。在新生儿持续性PAH患儿中,静脉注射西地那非后可明显改善患儿的氧合指数。裘刚等[36]采用西地那非、妥拉苏林、米力农对45例新生儿持续性PAH患儿治疗,结果显示,西地那非对新生儿持续性PAH具有良好的治疗效果,可有效降低mPAP。先天性心脏病术后PAH患儿能很好地耐受西地那非,无不良反应或只有轻度低血压[37]。于亦华[38]应用西地那非治疗18例先天性心脏病合并重度PAH患儿,结果显示,治疗有效率为89%(16/18)。有研究表明,西地那非对肺血管扩张的作用随着时间延长而减弱,后期如果想要维持类似的治疗效果,需不断增大药物剂量。考虑药物不良反应会随着剂量的增加而逐渐突出,目前欧美等国家并不推荐大剂量使用西地那非治疗儿童PAH(特别是17岁以下儿童)[39]。同时我国也缺少大样本、多中心的临床研究评估西地那非的有效性和安全性,故我国的西地那非说明书中并未将儿童PAH的治疗纳入其中。虽然西地那非已经广泛应用于儿童PAH的临床治疗,且很多临床研究提示其对儿童PAH的治疗效果较好,但仍然是一种超说明书用药,不受法律法规的保护。
治疗儿童PAH时常需要联合用药,不同药物作用的靶点通路不同,联合用药增加治疗效果的同时也减少了耐药的发生。2015年欧洲心脏病学会/欧洲呼吸病学会(ESC/ERS)发布的《PH诊断和治疗指南》[40]提出,PAH患儿的治疗需要有一套完整的策略,不仅是局限于单药治疗,根据对患儿心功能及PAH严重程度的评估,推荐心功能Ⅱ~Ⅲ级患儿采用联合药物治疗。LAJOIE等[41]2016年发表的一篇荟萃分析表明,联合用药较单药治疗PAH患儿的临床不良事件发生率可降低35%。美国FDA 2016年也颁授了PAH靶向药物的联合治疗方案:治疗病情较为严重的PAH患儿建议联合用药,包括:ERA+PDE-5抑制剂、PGI2+PDE-5抑制剂,其中mPAP<40 mm Hg,PVR<560 dyn.s/cm2时单用PDE-5抑制剂或ERA;41 mm Hg<mPAP<55 mm Hg,561 dyn.s/cm2<PVR<800 dyn.s/cm2时给予PDE-5抑制剂或ERA或PGI2双联治疗;mPAP>55 mm Hg,PVR>800 dyn.s/cm2时 给 予 PGI2和 /或PDE-5抑制剂和/或ERA;联合治疗后若效果仍不理想,可考虑三联治疗或肺移植等[42-43]。
4 小结
儿童PAH早期诊断及治疗非常关键,因其血流动力学特点及血管发育情况与成年人有着明显差别,并不能将成年人标准套用于儿童。目前各心血管中心对儿童PAP的靶向治疗还是以经验治疗为主,缺少行业统一标准,因此制定出儿童靶向治疗共识或指南十分必要。随着新的治疗思路的引入及靶向治疗药物在我国儿科临床中应用经验的积累和大样本长期随访研究的不断涌现,相信在不久的将来我国儿童PAH的靶向治疗会越来越规范。
本文无利益冲突。
猜你喜欢
临床肝胆病杂志(2022年6期)2022-11-25
肝博士(2022年3期)2022-06-30
保健与生活(2022年11期)2022-06-09
家庭医药(2022年2期)2022-02-18
食品安全导刊(2021年20期)2021-08-30
天津医科大学学报(2021年4期)2021-08-21
现代临床医学(2021年2期)2021-03-29
中西医结合肝病杂志(2020年2期)2020-10-27
中国实用医药(2017年3期)2017-03-20
安徽医科大学学报(2015年9期)2015-12-16
