
DDOD综合征ATP6V1B2基因新发变异及遗传学分析△
2020-07-27 08:31梁宾荣伽玲张晓康何思颖向阳罗晶张元珍马建鸿杨国华郑芳
听力学及言语疾病杂志 2020年4期
梁宾 荣伽玲 张晓康 何思颖 向阳 罗晶 张元珍 马建鸿,3 杨国华 郑芳,3
DDOD综合征(dominant deafness and onychodystrophy syndrome, OMIM#124480)又称先天性耳聋伴甲发育不全综合征,是一种罕见的先天性异常疾病,该病的主要临床特征是严重的感音神经性聋和指甲发育不全或缺失[1]。1961年,Feinmesser等首次报道了一个表现为耳聋、甲发育不全患者的家系,先证者为10岁女性,先天性聋,手指甲和脚趾甲营养不良且发育不全;其妹妹也有同样的表型,即双侧感音神经性聋伴甲发育不全[2]。随后,Kondoh[3]和White[4]等发现该病为常染色体显性遗传。2014年,通过对两个DDOD综合征家系全外显子测序,Yuan等[5]率先发现ATP6V1B2基因突变为DDOD综合征的致病原因。
本研究通过对一例先天性感音神经性聋伴指甲和牙齿发育异常的患者外周血进行二代测序,确定其致病突变位点并进行遗传学和生物信息学分析,为该家系的遗传咨询提供依据。
1 资料与方法
1.1研究对象 患者,女,6岁,出生时发现听力异常,2013年5月外院听性脑干反应(ABR)测试显示双耳反应阈值为99 dB nHL,诊断为重度感音神经性聋,人工耳蜗植入术后听觉语言康复效果良好。于2019年7月来武汉大学中南医院就诊,查体发现先证者左右手拇指发育不对称、牙齿部分发育异常(图1),父母听力及表型均正常。本研究经武汉大学中南医院医学伦理委员会通过并获得患者及家属知情同意。

图1 先证者牙齿发育异常及左右拇指发育不对称
1.2主要仪器及试剂 普通PCR仪 (Bio-Rad, USA);外周血DNA提取试剂盒(OMEGA BIO-TEK);PCR扩增试剂盒(Thermo, EU);PCR引物(天一辉远生物科技有限公司)。
1.3外周血基因组DNA提取 于2019年7月在武汉大学中南医院采集先证者及其父亲、母亲的空腹静脉全血2 ml,EDTA-K2抗凝。按照外周血DNA提取试剂盒说明书操作,提取外周血DNA并进行纯度测定,保证DNA纯度良好(A260/280nm:1.8~2.0,A260/230nm:1.9~2.5),将提取的DNA置于-20 ℃保存。
1.4二代测序 将先证者及父母基因组送至上海韦翰斯生物医药科技有限公司,通过探针杂交捕获558个耳聋相关致病基因全部外显子及毗邻剪接区域(约50 bp),并进行富集。对富集的基因进行质量控制,利用高通量测序仪(Illumina HiSeq X)进行测序。测序得到的原始数据首先去除不符合质控要求的reads,然后运用BWA软件与UCSC提供的hg19版本人类基因组参考序列进行比对,通过OMIM、ClinVar、HGMD等数据库对可疑突变进行相关注释,最终得到候选致病突变。
排除其他候选致病突变:先证者ESPN、USH2A基因上各发现一个杂合错义突变,ESPN基因异常可导致常染色体隐性遗传性聋36型;USH2A基因异常可导致常染色体隐性遗传的视网膜色素变性39型或Usher综合征IIA型,Usher综合征IIA型的主要临床表现为:出生时发生的感音神经性听力损失和进行性视网膜色素变性。ESPN、USH2A基因异常所致疾病与先证者临床表现相关,但常染色体隐性遗传病只检测到先证者的一个杂合变异,其父母未检测到,与遗传方式不符合,故排除。
1.5Sanger测序验证 用NCBI Primer-BLAST工具在线设计ATP6V1B2可疑致病突变位点c.1517G>A (p.R506Q)的引物,引物由天一辉远生物科技有限公司合成。引物序列如下:正向:CATCCTCTCCCGCAAGACTG,反向:ATAGCACTGAGGACCCAGGT。PCR扩增体系为50 μL,包括10×Taq DNA buffer 5 μL,25 mmol/L MgCl23 μL,10 mmol/L dNTPs 1 μL,Taq DNA聚合酶2 μL(1 U/μL),ddH2O 30 μL,10 μmol/L正反向引物各2 μL,模板DNA 5 μL(30~60 ng/μL)。扩增条件如下:95 ℃ 5 min;95 ℃ 30 s,60 ℃ 30 s,72 ℃ 1 min, 扩增35个循环;72 ℃ 10 min;4 ℃保存。PCR扩增产物经1%琼脂糖电泳验证条带单一且无外源DNA污染后,将扩增产物送至擎科伟业生物技术有限公司进行Sanger测序。
1.6生物信息学分析 用UniProt(https:∥www.uniprot.org/)在线工具分析ATP6V1B2错义突变位点在不同物种蛋白质间的序列差异,以评估该突变位点处氨基酸的进化保守性。用Mutation taster(http://www.mutationtaster.org/)在线工具预测错义突变位点的潜在致病性。用PolyPhen-2(http://genetics.bwh.harvard.edu/pph2/)在线工具预测错义突变位点对蛋白质结构的改变和对其功能的影响。用ANTHEPROT 6.9.3软件分析错义突变对ATP6V1B2编码蛋白质的二级结构和理化性质的影响。ANTHEPROT 6.9.3软件蛋白质二级结构示意图中,蓝色模块表示α-螺旋,红色模块表示β-折叠,白色模块代表其他无规则松散结构。蛋白质理化性质示意图中,横轴代表氨基酸序列,纵轴代表氨基酸亲水性数值大小。
2 结果
2.1遗传学分析结果 对先证者NGS结果分析显示在遗传性耳聋相关致病基因中,ATP6V1B2基因上有一个杂合突变位点c.1517G>A (p.R506Q)(图2a)。一代验证结果显示先证者父母均未检出该变异(图2b、c),推测该变异为新发变异,但不能排除父母一方存在生殖细胞嵌合的可能。将该变异与人类基因变异数据库(HGMD)、NCBI-Clinvar数据库和六万人外显子组整合数据库(ExAC)进行比对,均未发现该突变位点的报道。
2.2生物信息学预测结果 用UniProt比对ATP6V1B2编码的蛋白在人类、小鼠、大鼠、马等多个物种中氨基酸的序列,结果显示ATP6V1B2c.1517位点处的氨基酸在物种进化中高度保守(表1)。用PolyPhen-2对突变位点进行致病性预测,结果显示ATP6V1B2c.1517G>A(p.R506Q)为致病性突变,综合得分为1.00,提示该突变位点对蛋白质的功能有较大影响(图3a);用Mutation taster对PolyPhen-2的预测结果进行辅助验证,结果一致(图3b)。用ANTHEPROT 6.9.3软件对该基因编码的野生型和突变型蛋白质进行比对分析,结果显示ATP6V1B2p.R506Q突变可引起蛋白质二级结构的改变,且可使该位点氨基酸的亲水性增加(图4)。
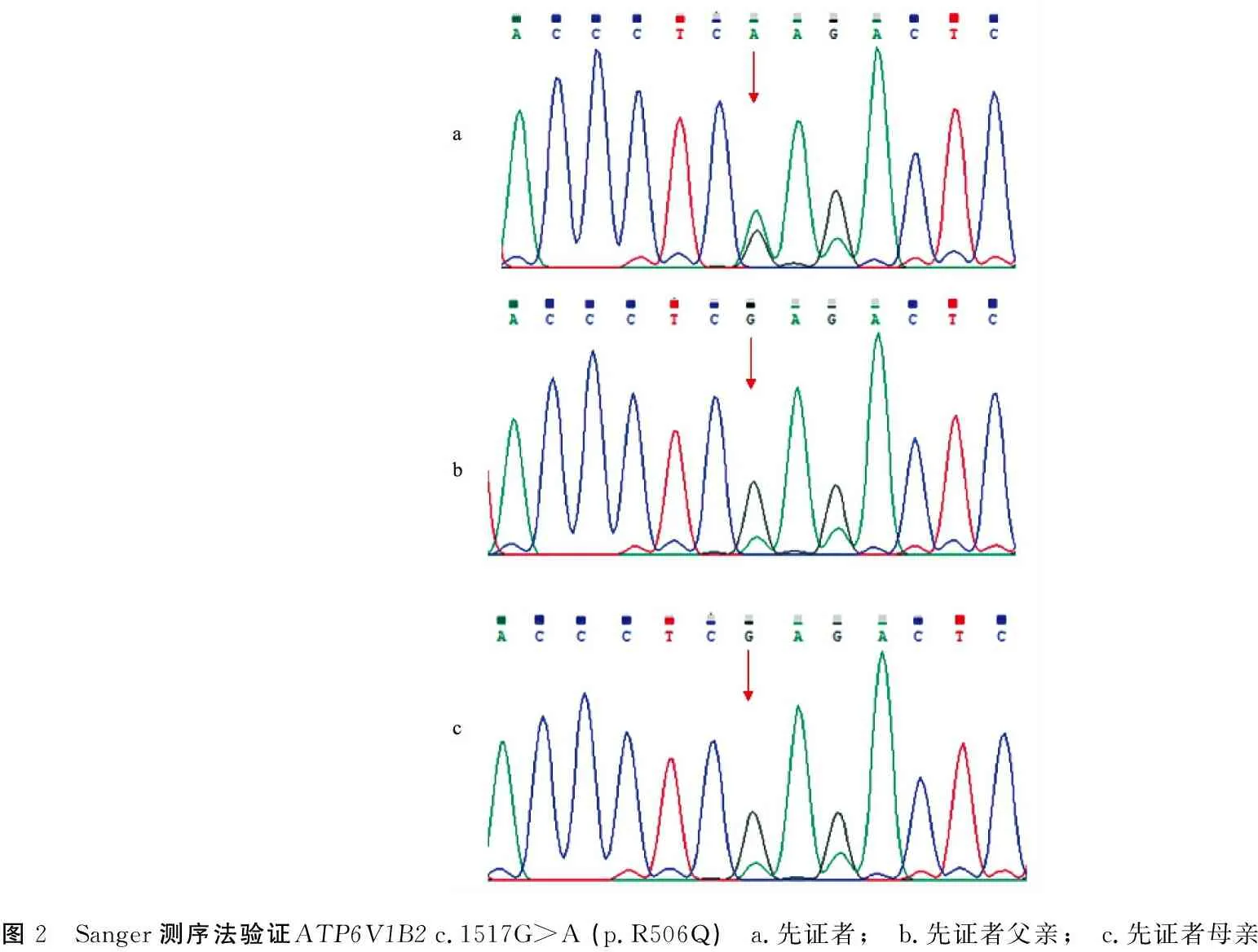
图2 Sanger测序法验证ATP6V1B2 c.1517G>A (p.R506Q) a.先证者; b.先证者父亲; c.先证者母亲

表1 ATP6V1B2c.1517(p.506)多物种序列比对
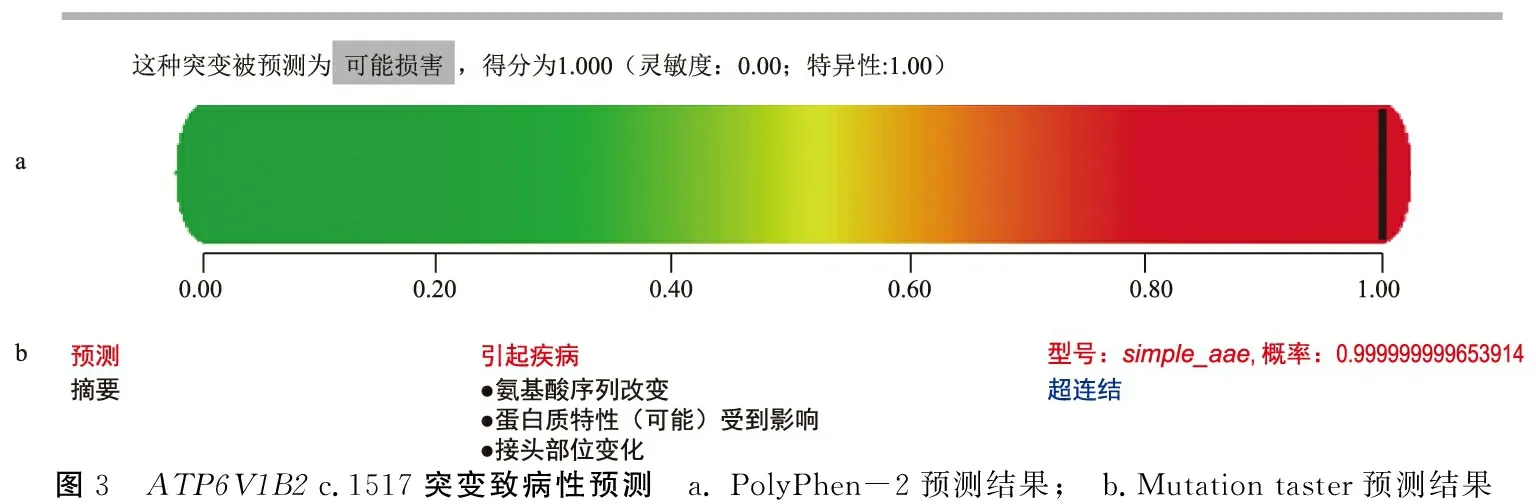
图3 ATP6V1B2 c.1517突变致病性预测 a. PolyPhen-2预测结果; b.Mutation taster预测结果

图4 ATP6V1B2 c.1517突变影响蛋白质二级结构和理化性质 a、b分别为野生型和突变型ATP6V1B2 c.1517附近蛋白质二级结构示意图; c、d分别为野生型和突变型ATP6V1B2 p.506氨基酸亲水性分析图(红线处指示突变位点)
3 讨论
DDOD综合征是一种罕见的常染色体显性遗传病,又称耳聋-甲发育不全综合征,主要表现为先天性感音神经性聋、甲发育不全、牙齿圆锥形伴间断缺失。迄今为止,全球共报道了11例DDOD综合征患者[6],其中只有4例有明确的致病基因,均为ATP6V1B2c.1516 C>T (p.Arg506X)突变[5, 7]。ATP6V1B2是DDOD综合征的主要致病基因,ATP6V1B2基因定位于染色体8p21.3,该基因总长度为2.875 kb,包括14个外显子,编码511个氨基酸。ATP6V1B2编码囊泡型质子泵蛋白 (V-ATPase)中的B2亚基。V-ATPase是一种由多亚基组成的质子泵,水解ATP逆浓度梯度转运H+[8],通过酸化细胞器参与体内多种重要的生理过程,如:蛋白的递呈、代谢物质的运输、受体调节的内吞作用等[9, 10]。V-ATPase至少由14个不同的亚基组成,分为负责催化ATP结合和水解的胞质水溶性V1区和负责质子转运的跨膜疏水性的V0区,其中V0区至少由a、d、c、c’、c” 5个不同的亚基组成[11],而V1区至少由A、B、C、D、E、F、G、H 8个不同的亚基组成。ATP6V1B2基因是V-ATPaseV1区的组成单位,在溶酶体、高尔基体、分泌型囊泡等多种细胞器膜上均有表达,广泛分布在人体器官组织中,在维持人体细胞正常功能方面发挥重要作用[12]。
本研究对一例先天性感音神经性聋伴手指、牙齿发育异常的先证者进行遗传学筛查,发现该先证者存在ATP6V1B2c.1517G>A (p.R506Q)杂合突变,父母表型正常且未检出异常突变,通过检索HGMD等多个变异数据库及相关文献,确证该位点为ATP6V1B2c.1517G>A (p.R506Q)新发突变。但尚不能排除由于其父母生殖细胞嵌合所致的后代突变,即父母生殖细胞存在正常和突变两种类型,而父母的体细胞正常所以父母表型正常,而其后代继承了生殖细胞的突变也就是胚系突变而出现病理表型。
生物信息学分析显示,该突变位点具有高度的进化保守性。突变位点致病性预测结果显示,ATP6V1B2c.1517G>A (p.R506Q)为致病性突变,蛋白质结构功能预测分析显示,该突变会引起囊泡型质子泵蛋白二级结构及亲水性发生改变,提示有可能是由于突变引发该蛋白质的二级结构和理化性质改变造成囊泡型质子泵蛋白的功能异常,从而对DDOD综合征的发病产生影响。2014年Yuan等[5]发现ATV6V1B2基因第14外显子相邻的位点存在另一种变异ATP6V1B2c.1516 C>T (p.Arg506X),该变异会导致溶酶体中的V-ATPases功能异常和酸化异常,并且应用斑马鱼模型证实了ATP6V1B2变异导致DDOD综合征的甲发育不全和指骨发育异常的表型。
值得注意的是,ATP6V1B2基因的致病变异不仅可以引起DDOD综合征,ATP6V1B2c.1454 G>C (p.Arg485Pro) 还可以造成Zimmermann-Laband 综合征 2 型(ZLS2) (OMIM#616455)的发生[13]。ZLS2是一种罕见的以面部发育异常伴有早期口内弥漫性牙龈纤维增生为特征的综合征。ZLS2患者通常表现出鼻突出,耳垂厚软,指甲、远端指骨发育不良或缺失[14]。本研究中的患者没有出现ZLS2的典型临床表现如弥漫性牙龈纤维增生、颅颌面部畸形,但符合远端指骨发育不全这一特征。然而在什么情况下,ATP6V1B2基因的致病变异导致DDOD综合征或ZLS2综合征,目前尚不清楚。但已报道的致DDOD综合征的突变ATP6V1B2c.1516 C>T (p.Arg506X)会插入一个过早的终止密码子,使得蛋白被截断,导致溶酶体的酸化功能异常。而致ZLS2综合征的突变ATP6V1B2c.1454 G>C (p.Arg485Pro)是错义突变,精氨酸被脯氨酸替换可能会通过破坏B亚基而扰乱V1亚基复合体内各亚基间的相互作用而产生更加严重的表型。
综上,本研究首次发现ATB6V1B2基因第14外显子存在一个新变异ATP6V1B2c.1517G>A (p.R506Q),结合患者典型的DDOD综合征的临床症状及生物信息学分析,推测该变异可能是其表型异常的主要原因,为该患者及其家系的遗传咨询提供了有力的分子依据。
猜你喜欢
中华医学图书情报杂志(2022年1期)2022-11-18
中国现代医生(2022年21期)2022-08-22
临床输血与检验(2022年3期)2022-06-22
麦类作物学报(2022年3期)2022-05-19
临床与实验病理学杂志(2022年3期)2022-04-06
实用肝脏病杂志(2022年2期)2022-03-21
西北农业学报(2021年4期)2021-05-19
天津医科大学学报(2021年1期)2021-01-26
诊断学(理论与实践)(2020年1期)2020-04-28
三农资讯半月报(2020年2期)2020-03-09
