
A Case Study on the Mechanisms of the Nucleation andGrowth of Silicene on the Surface of Calcium Silicate Hydrates
2020-07-24 08:30:46ZHOUYangZHENGHaojieQIUYuwenZOUXixiHUANGJiale
中国材料进展 2020年6期
ZHOU Yang, ZHENG Haojie, QIU Yuwen, ZOU Xixi, HUANG Jiale
(1. School of Materials Science and Engineering, Southeast University, Nanjing 211189, China) (2. State Key Laboratory of High Performance Civil Engineering Materials, Jiangsu Research Institute of Building Science Co., Ltd., Nanjing 211103, China)
Abstract:Silicene, a newly emerging two-dimensional (2D) material, can be a superior reinforcement candidate to enhance the mechanical performance of calcium silicate hydrates (C-S-H, the primary hydration product of portland cement), in consideration of the common substrate atom Si. Here, with the reactive molecular dynamics simulations, the possibility of depositing such a silicene monolayer on the C-S-H surface was investigated. The results indicate that high-reactivity non-bridging oxygen atoms (NBOs) on the C-S-H surface facilitates the nucleation of silicon atoms, by the formation of high-strength Si—O—Si bonds. The nucleated silicon clusters follow a relatively uniform distribution over the whole surface. As the deposition continues, clusters grow larger, leading to the merging and coalescences of neighboring ones, via the formation of bridging bonds and the transformation of cluster shape. Eventually, a relatively complete silicene monolayer has been successfully established on the C-S-H surface, with high surface coverage ratio. Furthermore, it suggests the deposition rate and temperature may substantially affect the nucleation and growth process of silicene. This study opens the perspective of depositing a 2D material on an inorganic matrix and elucidates the atomic stacking mechanism on the interfaces.
Key words:silicene; calcium silicate hydrates; molecular dynamics; nucleation; growth
1 Introduction
Over the past decade, silicene has become a new member of the two-dimensional (2D) materials family and attracted increasing attention because of the outstanding electronic and mechanical performance[1-3]. Silicene owns a similar structure with graphene except for a low-buckled geometry, which originates from the orbital hybridization of Si atoms which prefer sp3hybridization over sp2hybridization due to the greater Si—Si bond length. Although the in-plane Young’s modulus of silicene is relatively lower than graphene, previous researchers have indicated that the buckled structure of silicene dramatically increased its out-of-plane bending rigidity, even higher than that of graphene[1, 4-6]. This feature enlarges the application field for silicene in addition to electron devices[7].
One potential application of silicene is to relieve the brittleness of calcium silicate hydrates (C-S-H), which is the main hydration product of ordinary portland cement. As a main material of the basic building block, C-S-H is the strength source of concrete, which determines the service safety of global infrastructures[8-10]. Previous studies indicate that C-S-H is an amorphous phase while it exhibits a layered structure at nano-scale[11, 12]. The presence of large numbers of defects and pores in the interlayer region of C-S-H causes a weak cohesion between neighboring calcium silicate sheets, which in turn limits the mechanical performance at macro-scale[13-15]. In consideration of the high intrinsic plasticity and chemical compatibility with the C-S-H matrix[3, 4, 16], silicene can be a proper candidate to be intercalated into the interlayer of C-S-H, for the purpose of toughness enhancement. However, due to the high chemical reactivity that makes it unstable in air[16], there is as yet few experimental reports for the silicene monolayer. Hence, firstly, we need to theoretically evaluate if a single silicene sheet can be deposited on the substrate of C-S-H, and elucidate the interaction mechanisms between the silicene and C-S-H substrate.
Molecular dynamics, a numerical computation method based on force fields, can be a good alternative in predicting the physical and chemical behaviors at an atomistic scale. So far, molecular dynamics simulations have been widely applied onto the modeling of structure, composition, mechanical performance, and interfacial properties of C-S-H and other cementitious materials[17-19]. Pellenqetal.[15]proposed the first molecular model of C-S-H using molecular dynamics simulations according to the small angle neutron scattering (SANS) results[14], and calculated the mechanical properties, which were consistent with the experimental results. Gengetal.[20]concluded that the calcium to silicon ratio strongly affected the bulk modulus of C-S-H when subjected to a hydrostatic pressure. The tensile loading tests were also simulated by molecular dynamics and C-S-H exhibited a highly anisotropy. It suggested that C-S-H possessed the lowest ductility and strength along the interlayer arrangement direction, due to the relatively weak connections between neighboring calcium silicate sheets[21]. The intercalation of nano-reinforcements (polymers, graphene, 2D-silica) into the interlayer of C-S-H may significantly improve the mechanical performance of matrix since those additives filled in the defects and pores[21-23]. Furthermore, Zhouetal. employed molecular dynamics to investigate the interfacial mechanisms between C-S-H and organics with different functional groups. The results implied that the high-polarity carboxyl groups could tightly adsorb on the C-S-H surface due to the formation of strong ionic bonds, while no remarkable interactions were observed between C-S-H and the carbon backbone[13]. In this scenario, silicene, which has the same primary element with C-S-H, may form stronger connections with the cementitious matrix materials.
Therefore, for the first time in this work, molecular dynamics simulations were employed to explore the possibility of the nucleation and growth process of silicene monolayer sheet on the C-S-H substrate. The (001) plane was selected as the most probable surface for deposition. The effects of the deposition rate and temperature on the nucleation and growth of a silicene monolayer were also investigated.
2 Computational Methods
2.1 Force fields
The reactive force field ReaxFF was employed to simulate the nucleation and growth process of a silicene monolayer on the C-S-H substrate. The ReaxFF force field (C/O/H) was invented by Van Duin, originally applied in the large scale reactive chemical systems of hydrocarbons[24], and then developed to describe silica-water interfaces and calcium silicate hydrates (Ca/Si/O/H)[25-27]. With a bond order-bond distance scheme, the ReaxFF could monitor the breakage and formation process of chemical bonds and elucidate the mechanism of chemical reactions[28]. Since all the parameters are determined from the quantum chemical calculations, ReaxFF owns a higher accuracy than other empirical force fields,e.g. ClayFF, CSHFF, CVFF. The polymerization and rearrangements of silicate tetrahedra have been successfully captured by the ReaxFF force fields when the C-S-H matrix was subjected to a uniaxial tensile loading. Furthermore, utilizing the ReaxFF force field, Zhouetal. observed the formation of bonds between C-S-H atoms and nano-reinforcements (2D-silica), which also implies the applicability of ReaxFF in this system[29].
2.2 Computational model
A tobermorite 11 Å crystal structure[30]was selected as the base-model, since tobermorite is a C-S-H analog with a well-organized atomic arrangement. As shown in Fig.1a, the base-model has a layered structure, with calcium silicate sheets alternately aligned along the direction of [001] (zaxis). Within a sheet, silicate tetrahedra of infinite length are arranged in order. Large numbers of defects and pores are present in the interlayer region between neighboring sheets, where there is high possibility for the occurrence of silicene nucleation and growth. To construct a deposition surface, the base-model was cleaved along at the (001) plane (the center of the interlayer space) and the upper part was removed, leaving a 1 nm-thick substrate and 1 nm-thick vacuum slab (Fig.1b). With respect to the in-plane size, the computational model has a length ofx=8.82 nm,y=8.76 nm with periodic boundary conditions applied alongxandydirections (Fig.1c). A fixed non-periodic boundary was employed alongzdirection, while a reflecting wall was placed at the upper bound of the simulation cell to inhibit the escape of any atoms.

Fig.1 Schematic illustration of C-S-H substrate: (a) the original model of tobermorite, (b) the computational model, (c) top view of the (001) surface (green balls denote calcium ions, red and yellow balls indicate the oxygen atoms and sliicon atoms in the silicate tetrahedra, respectively)
2.3 Computational details
The model establishment was implemented in Materials Studio 7.0 and the molecular dynamics simulations were carried out using software LAMMPS[31], both on the platform in the High-Performance Computing Centre of Nanjing University. The ReaxFF force field was employed, with a Nose-Hover thermostat and a time step 0.25 fs to integrate the Verlet equations of motion. Firstly, the substrate slab was thermalized for 100 ps under canonical ensemble (NVT) at the temperature of 300 K. Subsequently, the deposition of silicene was simulated by randomly adding silicon atoms onto the C-S-H surface, highly similar to the experimental growth process of silicene (Si evaporation from a silicon crystal in ultra-high vacuum). It was guaranteed that each newly added Si atom was at least 2 Å away from already existing atoms in the system. During the deposition process, the atoms in the two bottom sheets of C-S-H were fixed to simulate the underlying bulk substrate, while the rest C-S-H atoms still experienced a NVT ensemble. In addition, a microcanonical ensemble (NVE) was applied onto the added silicon atoms.
The influence of deposition rate and temperature on the nucleation and growth process of silicene was also investigated. In consideration of the huge computational cost induced by reactive molecular dynamics simulations, three relatively high deposition rates were chosen:400, 1300, and 4000 atoms/ns. Besides, according to the real experimental conditions[32]and computational limits of molecular dynamics simulations, three different temperatures of 300, 450 and 600 K, were selected to examine the temperature independence. In each case, the trajectories of all atoms in the system were sampled every 25 fs to capture reaction details for analysis.
3 Results and Discussions
The number of generated silicon islands on the C-S-H surface under the deposition rate of 4000 atoms/ns at 300 K is illustrated in Fig.2, which implies three main stages during the formation process of a single silicene sheet. At stage I, the number of islands keeps going up, which suggests more and more isolated clusters are formed on the surface of C-S-H, corresponding to the nucleation process. At stage II, the number of islands experiences a downwards trend, since the clusters grow larger and neighboring ones begin to converge. Finally, at stage III, the number of islands maintains at an extremely low value, which indicates that the silicene monolayer has been formed. From a visual perspective, the atomistic trajectories during the entire process are recorded and the representative snapshots of each stage are illustrated in Fig.3. The atomistic configurations and transformation features during each stage will be presented in details in the following sections.

Fig.2 The number evolution of islands during the simulation process at 300 K with the deposition rate of 4000 atoms/ns

Fig.3 Schematic illustration of the Si—O—Si bonds on the interface of silicene and C-S-H (the red and yellow balls in blue background denote the C-S-H substrate, while yellow balls in white background denote the deposited silicon atoms)
3.1 Nucleation
The key problem is whether silicon atoms have a good affinity with the C-S-H surface, which determines the possibility of silicene nucleation. The non-bridging oxygen atoms (NBOs) at the surface of C-S-H, proved by previous studies[13, 21, 22]to possess a high polarity and reactivity, play a significant role. As shown in Fig.3, the deposited silicon atoms can form strong covalent bonds Si—O—Si with NBOs. These strong connections stabilize the adatoms and lead to the nucleation. Actually, large numbers of reactive Si—O groups are present at the C-S-H surface, which provide abundant deposition sites for silicon atoms. Besides, intrinsically, the formed Si—O—Si bonds are the basic components and strength source of C-S-H, which connect the silicate tetrahedra and constitute the skeleton[14, 15]. Therefore, the nucleation of silicon atoms at the surface of C-S-H is thermodynamically favorable.
At stage Ⅰ, a scattered nucleation of silicon atoms can be observed, as shown in Fig.4a. The new adatoms tend to be evenly distributed on the C-S-H surface instead of local concentration, forming tremendous islands, which correspond to the rapid increase of clusters (stage I in Fig.2). Locally, the formation process of a typical nucleated cluster is illustrated in Fig.4d~4i. During this stage, neighboring atoms connect with covalent bonds, forming a small cluster. It grows larger like a branch as more atoms are deposited onto the surface. Once the boundary atom of this cluster is adequately close to other clusters, the coalescence of two independent islands may occur (see Fig.4h and 4i). Overall,the surface coverage ratio at this stage is relatively low, while clusters have a small size and large number.

Fig.4 Schematic illustration of nucleation and growth process of a silicene monolayer (a~c); the nucleation of a small cluster in Fig.4a (d~i); the growth process of a cluster in Fig.4b (j~m)
It is suggested that silicon atoms should exhibit a ring structure as depositing on a defect free Ir surface[33], however, here the newly nucleated silicon clusters tend to form a branch structure. The nucleation mechanisms may also explain the difference. As mentioned above, it is the reactive Si—O sites on the substrate that attract the nucleation of silicon atoms. Since the NBOs are almost aligned in a line on the C-S-H surface, the nucleation of silicon atoms also follows the line.
3.2 Growth
As the deposition of silicene atoms continues, clusters grow larger and the surface coverage ratio increases (see Fig.4b). During stage Ⅱ, the dominant shape of islands transforms from branch to polygon ring. This is because the new silicon adatoms are attracted by the existing silicon atoms, forming the Si—Si connections, regardless of the original site preference. The ring structures of silicon atoms may have 4~7 members, dependent on the local chemical environment. They may also change the shape and member size of polygons to accommodate new atoms. During 12.125~12.800 ps (see Fig.4j and 4k), a five-member-ring (marked by green dashed circles) recruits two adatoms to become a seven-member-ring contemporarily. However, shortly, one atom of this seven-member-ring is grabbed by a neighboring ring structure, due to the thermodynamic instability (at 13.275 ps, Fig.4l). Hereinafter, the six-member-ring may last for a relatively long time since it is a defect free structure compared with a five/seven-member-ring[33].
Furthermore, the clusters grow larger, leading to substantial surface coverage, thus neighboring islands become to converge by the formation of bridging bonds. At 12.125 ps (Fig.4j), the two clusters in each dashed circle are isolated, while at 12.800 ps (Fig.4k), the coalescence of islands (marked by red dashed circles) occurs due to the deposition of two atoms on the boundary. The two adatoms form bonds with each island and act as a bridge connecting them together. Another coalescence mechanism is the high-mobility of boundary atoms. Even though during 13.275~13.500 ps (see Fig.4l and 4m), no new atoms are deposited on the boundary between the left-part branch and right-part polygon cluster, the coalescence still takes place (marked by blue dashed circles). This may be attributed to the instability of four-member-ring, which increases the mobility of member atoms. In such a scenario, the ring can be open and the boundary atoms form a dangling bond with the branch cluster. As a consequence of these two coalescence mechanisms, the number of independent islands begins to decrease during this stage, as shown in Fig.2.
He drove twice round the city, throwing money to the right and left, and the third time, as he passed under the palace windows, he saw Ludovine lift a corner of the curtain and peep out
As the deposition proceeds, the cluster coalescence and growth continues until a complete monolayer of silicene is formed on the C-S-H surface, as shown in Fig.4c. Although the surface coverage ratio is relatively high, some defects still remain due to the low defect formation energy[34]. The large voids are generated by the non-uniform growth of silicon atoms. It is predicted that they can be eliminated at simulations with larger spatial or temporal scales, owing to the superior defect mobility of silicene. Furthermore, there are minor height deviations within the formed silicene surface, which neither exhibits an absolutely flat nor a buckling shape as suggested by density function theory (DFT) studies[1, 32]. It may be attributed to the uneven C-S-H surface.
3.3 The influence of deposition rate and temperature
The effects of deposition rate and temperature on the nucleation and growth process of a silicene monolayer are illustrated in Fig.5 and 6, respectively. It seems that a slower deposition rate contributes to a more uniform nucleation. At the rate of 4000 atoms/ns, the peak of the island number positions at around 400, while at the rate of 400 atoms/ns the peak value is almost the same in spite of a much larger atom number (around 650). As shown in Fig.6, the deposition temperature has a more remarkable effect on both the nucleation and growth process. Although peaked at the same position, the island number owns a smaller maximum value under higher temperature. It may be caused by the higher mobility of silicon atoms, which increases the likelihood of clusters contact and merging with each other. The same atom number and lower island number indicates a larger cluster size at higher temperature, which is consistent with other results[33]. These large clusters have a smaller specific surface area and lower possibility of contact with other clusters, which leads to a slower coalescence rate compared with small islands (see the decrease slope in Fig.6).
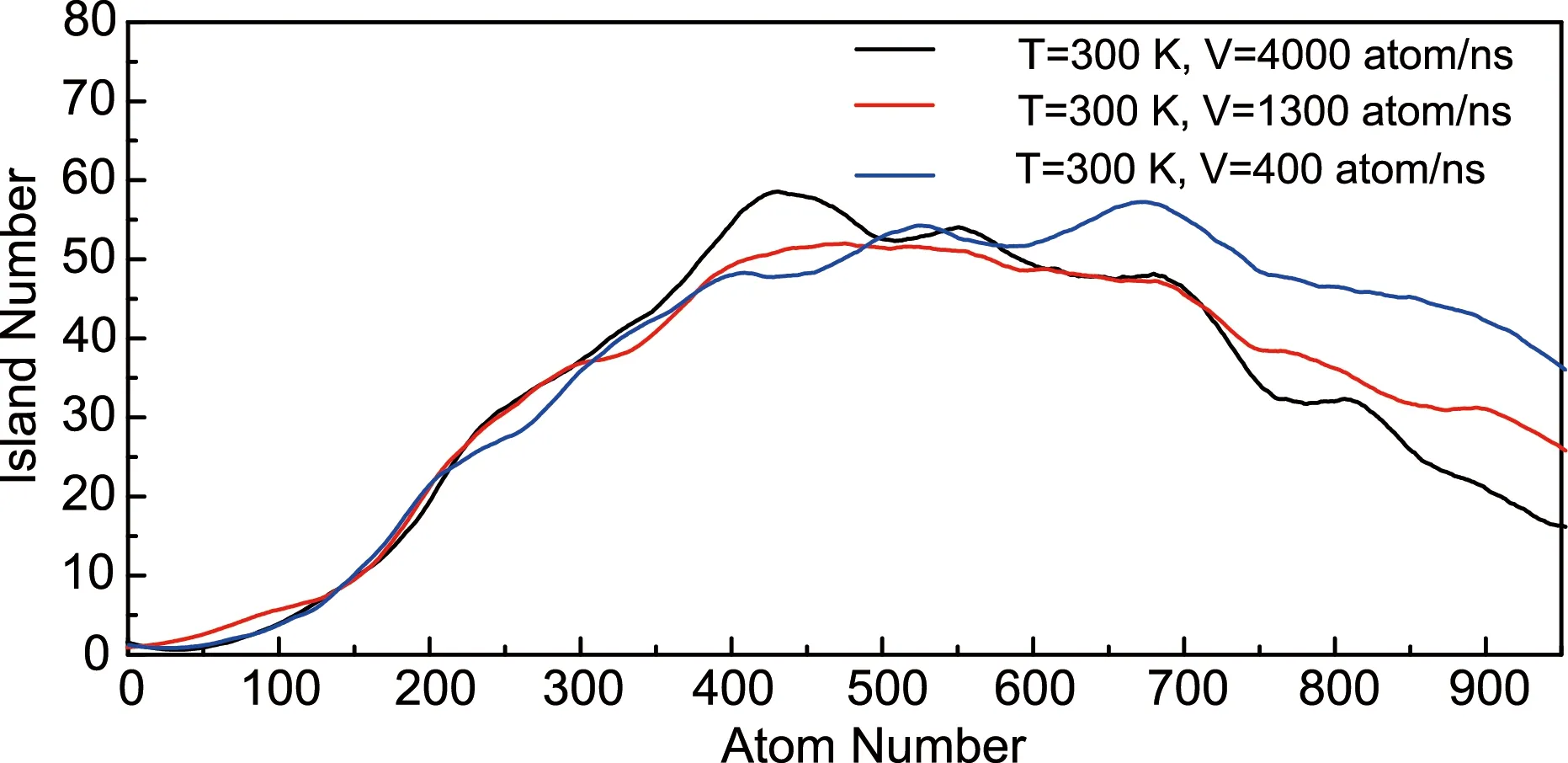
Fig.5 The number evolution of islands during the simulation process at 300 K with different deposition rates

Fig.6 The number evolution of islands during the simulation process with the deposition rate of 4000 atoms/ns at different deposition temperatures
4 Conclusions
C-S-H/silicene is a desired stratified nanocomposite to enhance the mechanical performance of C-S-H in consideration of the common substrate atom Si. In this study, the reactive molecular dynamics was employed to explore the possibility of depositing a silicene monolayer on a C-S-H surface (001).
The results indicate that the presence of large numbers of high-reactivity NBOs on the C-S-H surface provides abundant deposition sites for silicon atoms. The adatoms can form high-strength Si—O—Si bonds once connected with NBOs, which are also the intrinsic components and cohesion source of C-S-H skeleton. The nucleation follows a relatively uniform distribution over the whole surface. Within each small cluster, the deposited atoms exhibit a branch shape during the nucleation stage. As the deposition continues, more silicon atoms are connected with the existing islands, changing the size and shape of the original clusters. The ring structure becomes dominant during the growth stage, among which the six-member-ring is more thermodynamically stable. The growth of clusters causes the merging and coalescences of neighboring ones, by the formation of bridging bonds and the transformation of cluster shape.
Eventually, a relatively complete silicene monolayer can be successfully established on the C-S-H surface. However, some defects still remain,i.e. large voids and height deviations. The large voids are formed due to the non-uniform growth of silicon atoms, which can be eliminated by larger spatial- or temporal-scale simulations. Besides, out of the plane, the silicene monolayer exhibits irregular height deviations rather than an ideal buckling structure (as suggested by DFT studies). It should be attributed to the unevenness of depositional plane and further surface modifications on C-S-H are still needed. Furthermore, the deposition rate and temperature may substantially affect the nucleation and growth process of silicene. A slower deposition rate contributes to a more uniform nucleation, while higher temperature increases the size of clusters. This study opens the perspective of depositing a 2D material on an inorganic matrix materials and elucidates the atomic stacking mechanism on the interfaces, which is a potential measure to fabricate high performance cement-based materials.
