
Overview of genetic signaling pathway interactions within cutaneous malignancies
2020-07-20 03:48:10BrittanyManerLeonieDupuisAshleySuJeremyJuengTannerHardingJohnMeisenheimerVIIFahadSiddiquiMiaHardackSavinaAnejaJamesSolomon2
Brittany S.Maner, Leonie Dupuis, Ashley Su, Jeremy J Jueng, Tanner P.Harding, John Meisenheimer VII, Fahad S.Siddiqui, Mia R.Hardack, Savina Aneja,, James A.Solomon2,,-9
1Ross University School of Medicine, Miramar, FL 33027, USA.
2Ameriderm Research, Ormond Beach, FL 32174, USA.
3University of Central Florida College of Medicine, Orlando, FL 32827, USA.
4University of South Florida Morsani College of Medicine, Tampa, FL 33612, USA.
5Alabama College of Osteopathic Medicine, Dothan, AL 36303, USA.
6Advanced Dermatology and Cosmetic Surgery, Orlando, FL 32806, USA.
7Florida State University College of Medicine, Orlando, FL 32304, USA.
8Kansas City University of Medicine & Biosciences, Kansas City, MO 64106, USA.
9Carle-Illinois College of Medicine, Urbana, IL 61820, USA.
Abstract Melanoma and non-melanoma cutaneous malignancies are some of the leading causes of cancer-related death in the United States.Though melanoma is more known to have a high mortality rate, the total mortality per year is nearly equal for between melanoma and non-melanoma skin cancer.Moreover, the non-melanoma types of cutaneous malignancies have potential to become locally invasive and even metastasize with very little to no treatment options when advanced.The development of these malignancies involves various genetic pathways through the four hallmarks of cancer development: malignant cell growth, apoptosis evasion, the use of supporting stroma and vascularization, and modulating and promoting an inadequate immune response.The genetic signaling pathways of basal cell carcinoma, squamous cell carcinoma, verrucous carcinoma, basosquamous cell carcinoma,melanoma, and cutaneous T-cell lymphoma interact with each other through genetic predisposition as well as with environmental exposures.Furthermore, solar ultraviolet radiation and chronic inflammatory states are found to initiate the progression of many of these cutaneous malignancies.This paper includes validated models of genetic pathways, emerging pathways, and crosstalk between genetic pathways through the four hallmarks of cancer development.Moreover, unlike most reviews addressing oncogenetics of the well-recognized, as well as newlydiscovered, genetic pathway mutations, this review stresses that these pathways are not fixed but rather exist in dynamic, interrelated, interactive, complex, and adaptive flux states.
Keywords: Basal cell carcinoma, squamous cell carcinoma, verrucous carcinoma, basosquamous cell carcinoma,melanoma, cutaneous T-cell lymphoma, genetic pathways, cancer hallmarks
INTRODUCTION
The molecular and genetic basis of the development of cutaneous malignancies involve multiple pathways which continue to evolve over time.Tumor initiation, promotion, and progression leading to the multiplication of abnormal cells are all effected by genetics, environmental factors, acute and chronic exposures, diet, trauma, and many other factors.Angiogenesis and metastasis are likely influenced by host dependent factors such as age and immunological status.Many skin malignancies environmental factors,such as UV radiation that causes photocarcinogenesis, are coupled with genetic and epigenetic alterations or extrinsic factors that contribute to local inflammation and dysregulation of normal pathways.Emerging evidence supports the role of chronic inflammation in skin carcinogenesis mediated by factors including nuclear factor-kappa B (NF-κB), signal transducer and activator of transcription 3 (STAT3), and hypoxiainducible factor-1 alpha (HIF-1α).In this article, we review validated models including the hedgehog pathway for basal cell carcinomas, p53 (TP53) pathway for squamous cell carcinomas, and BRAF pathway for cutaneous melanomas, among others.Additionally, we address emerging pathways that have not been completely elucidated including those implicated in cutaneous T-cell lymphoma.We also review the presumptive biology of less common skin cancers including basosquamous cell carcinoma and verrucous carcinoma.
In this review we approach the role of genetic events in relationship to four interactive processes referred to as “cancer hallmarks”.These signals must interact for cells which have undergone a carcinogenic event to survive, proliferate, maintain a footing, and spread.The four hallmarks we review are: (1) malignant cell growth; (2) prevention of apoptosis; (3) promoting use of supporting stroma and vascularization;and (4) modulating and promoting an inadequate immune response.The genetic pathways of cutaneous malignancies will be grouped as related to basal cell carcinoma, squamous cell carcinoma, verrucous carcinoma, basosquamous cell carcinoma, melanoma, and cutaneous T-cell lymphoma.Nonetheless, this review differs from most discussions concerning oncogenetics because we stress that these well-recognized,as well as newly discovered, genetic pathway mutations are not fixed but rather exist in dynamic,interrelated, interactive, complex, and adaptive flux states.
GENETICS OF BASAL CELL CARCINOMA
Nonmelanoma skin cancers (NMSC) include cancers affecting keratinocytes such as basal cell carcinoma(BCC) and squamous cell carcinoma (SCC).This term also includes Merkel cell carcinoma (MCC), a rare neuroendocrine tumor, and cutaneous T-cell lymphoma (CTCL).Four interactive processes referred to as the four hallmarks must interact for cells which have undergone a carcinogenic event to survive, proliferate,maintain a footing, and spread.
Malignant cell growth
Ultraviolet radiation
The most prevalent carcinogenic event promoting NMSC is ultraviolet radiation (UV) from sun exposure and/or from other UV sources such as tanning beds[1].Skin damage from sun exposure modulates tumorigenesis by damaging DNA, creating an inflammatory state, and activating oxidative stress responses,receptor tyrosine kinases (RTK), and pro-apoptotic pathways[2].UV, a recognized carcinogen, penetratesthrough the skin and damages keratinocytes of the basal epidermal layer.UV is emitted from the sun as well as from some man-made light.UVA (315-400 nm) compared to UVB (280-315 nm) accounts for 95% of sunlight.UVA is the primary source of light in tanning beds and it is less mutagenic[3,4].UVA penetrates deeper into the epithelium affecting dermal stroma, while UVB is absorbed into the stratum corneum layer[5].Primarily, UV causes DNA damage by generating cyclobutene pyrimidine dimers and 6-4 photoproducts[6].Under stress, Sestrin2, an antioxidant, is activated by tumor suppressor p53 and inhibits positive cell growth regulator mammalian target of rapamycin (mTOR)[7,8].The activity and skin penetration of UV depends on its wavelength with UVA penetrating between 700 nm - 1 mm and UVB penetrating between 280-320 nm[9].
Damage to chromosome 9 hedgehog pathway
BCC is the most common NMSC and accounts for 80% of NMSC diagnoses[10].BCC-specific risk factors include intermittent/recreational sun exposure, other sources of UV light, ionizing radiation, and skin phototype[11].The most critical pathway in BCC tumorigenesis appears to be the Hedgehog [sonic hedgehog(SHH)] pathway composed of three genes:SHH, DHH,andIHHas well as twopatchedgenes:PTCH1andPTCH2.The SHH pathway is important for the patterning, growth, and development of vertebrates.Thepatchedgenes encode SHH pathway receptors responsible for suppressing Smoothened(SMO), a transmembrane protein/proto-oncogene capable of activating Glioma-associated oncogene (Gli)transcription factors[12].In the SHH pathway, a SHH ligand binds to its receptor, PTCH (a transmembrane protein) to disinhibit SMO[13].The activation of SMO, which occurs in the primary cilium, causes the accumulation of intracellular calcium ions resulting in a disruption in calcium homeostasis[14,15].The transcription of Gli proteins has been shown to be sufficient to induce BCC development[16].After the SHH pathway,TP53gene point mutations are the second most common genetic mutation in BCCs[11,17].Traditional advanced BCC therapy consists of surgical resection, but the advent of small-molecule inhibitors of the SHH pathway allowed for new therapeutic options for patients with locally advanced or metastatic BCC[13,18].
Activation of the SHH pathway at the level of PTCH involves SHH interaction with PTCH through two distinct interfaces, the interface between PTCH and the calcium and zinc binding surfaces of SHH and the interface between PTCH and theN-terminal palmitoyl andC-terminal cholesteroyl moiety of SHH.Mutations at these interfaces that increase SHH binding to PTCH may subsequently increase the signaling strength of the SHH pathway and may drive tumorigenesis in BCC.Activation of the SHH pathway at the level of SMO involves side-chain oxysterols (endogenous cholesterol metabolites) that induce SMO accumulation in primary cilia even in the absence of SHH ligands.Excess side-chain oxysterols may lead to overactivation of the SHH pathway while pharmaceutical and genetic approaches aimed at reducing cellular cholesterol levels have been shown to attenuate SHH signaling in target cells, highlighting a potential role of excess side-chain oxysterols in the pathogenesis of BCC[19][Figure 1].

Figure 1.Sonic hedgehog signaling pathway.This figure describes the genetic signaling pathway of SHH.SHH: sonic hedgehog; SMO:smoothened
DNA damage repair
Damaged keratinocytes must depend on DNA repair mechanisms such as ataxia-telangiectasiamutated (ATM) and ataxia telangiectasia and Rad3-related (ATR) proteins, members of the PIKK family of proteins.After the cell experiences a UV-induced double-stranded DNA break, ATM undergoes autophosphorylation which empowers ATM to phosphorylate checkpoint kinase (CHK) 2[20,21].CHK2 inhibits CD25 phosphatases which then prohibit the cell from undergoing mitosis[22].ATM also can phosphorylate p53, arresting the cell in G1[23].If the UV induces a single-stranded DNA break, ATR will be activated.ATR will autophosphorylate and then CHK1 which goes on to phosphorylate CDC25[24,25].This,once again, prevents the cell from completing mitosis.In addition, ATR is involved in the p21 pathway which can modulate various cyclins/CDKs to inhibit the cell cycle[26].ATR phosphorylates murine double minute 2 (MDM2) to then inactivate p53.The inactivation of p53 results in the uncontrolled proliferationof cells with damaged DNA.On the other hand, ATR and ATM can directly phosphorylate E3 ubiquitin ligase SIAH1 which activates p53.The activation of p53 sends cells that normally should be permitted to proliferate to go into apoptosis.Thus, the dysregulation of ATR and ATM results in unpredictable cell cycle activity, especially at the p53 checkpoint which is known as “the guardian of the genome”.
Apoptosis evasion
Role of UV
UV exposure activates stress responses in the epidermis such as reactive oxygen species production which damages lipids, proteins, and DNA while also invoking antioxidant defense by suppressing tumorigenesis and initiating apoptotic pathways[27].The conflicting evidence forces the consideration that antioxidative therapies may be counterintuitive in the treatment of certain cancers.When observing the tumors of melanoma and non-melanoma patients, NMSC had lower levels of superoxide dismutase and catalase when compared to the melanoma samples.This observation suggests that NMSC are associated with weaker antioxidative defenses during tumorigenesis[28].To examine the signaling involved in oxidative stress, we will explore the p38 mitogen-activated protein kinases (p38) and c-Jun N-terminal kinases (JNK) pathways,both of which have been shown to be involved in pro- and anti-apoptotic mechanisms[29].
P38 signaling
P38 is a Raf-mitogen activated protein kinase (MAPK) protein that can respond to oxidative stress by triggering apoptosis.When oxidative stress activates the p38 system in keratinocytes, apoptosis signalregulating kinase 1 may become activated[30].Oxidative stress also inhibits MAPK phosphatases resulting in increased p38 activation[31].UV alone suffices in activating the p38 signaling pathway[30,32].
JNK signaling
JNK is another member of the MAPK family that can be activated in as little as 5 min post-UV exposure.JNK targets activator protein-1 (AP-1) which is an oncogenic transcription factor involved in cell cycle regulation[33].Although associated with pro-apoptotic activity, AP-1 can promote survival through crosstalk with the NF-kB pathway.Pharmacological studies have shown that the inhibition of JNK in human keratinocytesin vitroresults in greater UV-induced apoptosis[34].The same effect was shownin vivowhen inhibiting AP-1 in dominant negative c-jun hairless mice[35].
Stroma vascularization effects
RTK activation
RTKs are a group of receptors activated in response to UV exposure[36].Some of the more well-known RTKs include the IGF1-R, EGFR, FGFR, and VEGFR[37].RTK activation and RTK downstream pathways are targets for drug development[38].In NMSC, cetuximab (an EGFR inhibitor) is available as a pharmaceutical therapy[39].There are two main RTK activation pathways in NMSC, PI3K/mTOR signaling and RAF/MEK/ERK signaling[37][Figure 2].
PI3K/mTOR signaling
There are two forms of mTOR, mTORC1 and mTORC2, and both forms are involved in the development of NMSC secondary to sun exposure[40].PI3K and tensin homolog (PTEN) mutations are common in NMSC[41,42].To activate mTORC1, PI3K is recruited to the receptor which then leads to the phosphorylation of the p85 subunit.This allows PI3K to phosphorylate PIP2, converting it to PIP3.PIP3 then recruits phosphoinositide-dependent kinase 1 and AKT to the receptor to activate AKT.AKT then phosphorylates the MTORC1 negative regulator which leads to the activation of mTORC1, signaling for cell growth and proliferation[43,44][Figure 3].

Figure 2.Receptor tyrosine kinase genetic signaling pathway.This figure shows the epidermal growth factor receptor/receptor tyrosine kinase pathways.PDGF: platelet derived growth factor; TGF: transforming growth factor
Activation of mTORC2 occurs once PI3K phosphorylates PIP2 to PIP3[45].Part of the SIN1 protein is recruited to the cell membrane where it binds to PIP3, resulting in a conformational change in mTORC2 that reveals an active kinase site[24].PIP3 then recruits AKT to mTORC2 which results in the phosphorylation of AKT at the active kinase site[46].
FOXO3 signaling
The FOX”O” family of proteins are transcription factors known to regulate tumor longevity and suppression[47,48].FOXO3a transcriptionally targets apoptotic genes such asFasL, TNF-related apoptosisinducing ligand gene (TRAIL), BIM, andPUMA, all of which are involved in UV-induced apoptosis[49-51].AKT is a negative regulator of FOKO3a that acts by translocating into the cell nucleus with a chaperone protein, 14-3-3[52].Together, AKT and its chaperone reduce FOXO3a activity, sensitizing keratinocytes to UVB-induced apoptosis[49-51,53].
Raf/MEK/ERK signaling
The Raf/MEK/ERK pathway can be activated secondary to UV exposure.Raf binds MEK1 and MEK2,phosphorylating them.MEK1 and MEK2 then phosphorylate ERK[54].ERK1 is involved in the regulation of cell growth, malignant transformation, and drug resistance[55].A study found that cells treated with SMO inhibitors (a hedgehog pathway BCC treatment) have a tendency to increase RAS/MEK/ERK signaling,leading to the development of SCC[56].Thus, therapies should consider targeting the mTORC2/AKT and RAF/MEK/ERK pathways to simultaneously suppress BCC and SCC malignancies[57].
Isoelectric signaling
There appears to be a relationship between the isoelectric signaling and gene modulation.One study showed that IFN-γ can lead to the loss of EGF’s inhibition of basolateral K+channel activity[58].IFN-γ levels are often elevated during periods of inflammation which leads to the shift in signaling to favor the EGF-stimulated pathways (such as Raf/MEK/ERK and PI3K/mTOR described above)[59].
Modifying immune response
NF-κB pathway
Aside from the direct effects of sunlight, skin injury resulting in inflammatory response may also lead to tumorigenesis in NMSC.When the skin is damaged, such as in UV-induced sunburns, the microenvironment of the damaged area changes to allow for the extravasation of leukocytes to the tissueinjury site.NFκB is an established pathway in the mediation of inflammation.NFκB is a heterodimer of p65 and p50 subunits who are both bound to IkB, an inhibitory protein.Toll-like receptors, tumor necrosis factor (TNF) receptors, and RTK can all cause NFκB activation by activating IkB via phosphorylation[60].This process removes the inhibition of NFκB, allowing the protein to move into the nucleus and promote the transcription of pro-inflammatory genes such as TNF-α, IL-1 IL-6, IL-8, and various other cytokines and interferons[61].Furthermore, p65-dependent NFκB signaling nurtures a pro-inflammatory environment inducing SCC tumor initiation and promotion[60,61].
STAT3 pathway
Pro-inflammatory growth factors and cytokines induce the expression of the STAT family of transcription factor.STAT3 enhances the transcription of factors related to inflammation, tumor promotion, cell survival,and metastasis[62].In mice exposed to UVB, the overexpression of STAT3 results in accelerated skin tumorigenesis whereas the removal of STAT3 genes confers a resistance to skin tumor formation[63].
Once thought of as independent determinants of tumorigenesis, the four hallmarks of cancer form an interdependent network of signals that promote successful tumor growth.The study of skin cancer occurs under various conditions and laboratory settings.The high degree of variability in models, UV dosage,dimensions (2Dvs.3D), and methodologies creates potential confounds in the comparison of cancer signaling pathway.
GENETICS OF SQUAMOUS CELL CARCINOMA
Cutaneous squamous cell carcinoma (cSCC) is one of the most common forms of skin cancer worldwide.cSCCs most commonly present on the head and neck, particularly on the forehead, face, ears, and cheeks.Although melanoma is categorically perceived as a higher threat in society, more than 15,000 patients die from metastatic cSCC each year in the United States; this figure exceeds the total mortalities attributed annually to melanoma[64].In addition, the mortality rate of metastatic cSCC is over 70%[65].While multiple treatment modalities have been developed for advanced melanoma, advanced cSCC continues to have poor prognosis and limit options for treatment besides a newer immune checkpoint inhibitor, Cemplimab[66].
The factors evoking the carcinogenesis of cSCC are multiple and varied; the primary contributor is exposure to solar UV radiation.cSCC may also be initiated by industrial and inorganic exposures to tar,crude paraffin oil, fuel oil, creosote, lubricating oil, nitrosoureas, and arsenic in medications, foods, and drinking water[67].Organ transplant recipients and other immunosuppressed patients have increased risk for cSCC, especially locally advanced cSCC (lacSCC) and metastatic cSCC.Additionally, cSCC can develop in the setting of chronic inflammation.For example, cSCCs may develop from chronic ulcers, psoriasis,cutaneous lupus erythematosus, radiation dermatitis, porokeratosis, and lichen sclerosus.Understanding the various genetic pathways in which cSCC emerges will provide insight into improved and individualized treatments of cSCCs.The underlying genetic processes involved in the initiation, promotion, maintenance,and establishment of aggressive growth patterns resemble the pathophysiology hallmarks common to all cancers: cell growth, prevention of apoptosis, development of supportive stroma and vascularization, and modulation of the immune response.
Thus, each of the carcinogenic pathways for cSCC will follow complex genetic crosstalk between pathways for each of these hallmark systems as well as between these components.These signaling pathways may be unique to a hallmark component of the genetic pathway or be present in multiple parts of the genetic pathway.These genetic pathways include primary, well recognized, genetic mutations of signaling pathways,initiation of alternative genetic pathways, or modulation to and between alternative genetic pathways.Each of these signaling pathways may vary in how it affects the overall status of cSCC through one or more hallmarks systems.
UV radiation
Cell growth
The EGFR pathway is important for keratinocyte proliferation, turnover, and wound repair.In normal cells,EGF, transforming growth factor-α (TGF-α), TGF-β, and platelet derived growth factor (PDGF) are four of many ligands that must bind to EGFR for its activation.After binding, the receptor undergoes dimerization and autophosphorylation, followed by the signaling proteins with the SH2 domain attaching to the Grb2 adapter protein and SOS complex[68].This cascade of events continues with phosphorylation of the Ras/Raf/Map kinase pathway which then activates mTOR pathways[69].This cascade of events is similar to that described in the BCC section.mTOR causes a cascade of signaling events that culminate in combinations of DNA replication, protein synthesis, and lipogenesis.Hyperproliferative lesions, including psoriatic plaques, seborrheic keratoses, and cutaneous malignancies, have been found to have mutations in EGFR[70].Often this is due to the uncontrolled activation of the receptor without the necessary ligands present (i.e.,EGF, TGF-α and β, PDGF).In addition, UV mutates the EGFR, promoting uncontrolled keratinocyte replication and eventually cSCC showing that these chronic inflammatory conditions also have malignant potential [Figure 2].The similarities in pathway mutations between BCCs and cSCC demonstrate that BCCs and cSCCs have the potential to have modulating and communicating pathways.Rarely discussed, is this concept that the homeostatic flux of signals is imbalanced, allowing malignant keratinocytes to behave like benign keratinocytes through the option to differentiate into a BCC or cSCC.
Genetic crosstalk
In addition, crosstalk between these genetic events plays a marked role in cSCC.The EGF ligand promotes the activation of the EGFR pathways stimulating wound repair and eventually promotes hyperproliferative disease if uncontrolled.In addition, the ligands that activate EGFR can also activate the SHH pathway.It has been found that some BCCs have the potential of mutating to SCCs.The exact pathophysiology behind this event will be discussed in the basosquamous cell carcinoma section.Given that the mutation of the EGFR signaling pathway is known for producing SCCs, this pathway also has crosstalk with the SHH pathway, recognized as a culprit for the growth of BCCs.The pathway has been shown to increase intracellular calcium through activation of protein kinase C and MMPs, which incites further EGFR pathway activation[71][Figure 4].However, the extracellular environment found within the mouse embryonic stem cells could be a hypercalcemic environment.A hypocalcemic environment may causean imbalance within the SHH pathway leading to atypical signaling and activation of other modulating receptors[71][Figure 5].Because the crosstalk between the EGFR pathway and SHH pathway occurs early in the activation of the SHH pathway, the blockade of SHH can lead to shunting of the SHH pathway to the EGFR pathway causing upregulation in the EGFR pathway and increased proliferation of keratinocytes.Eventually, these tumors have the potential to mutate to a different type of skin cancer [Figure 4][72].
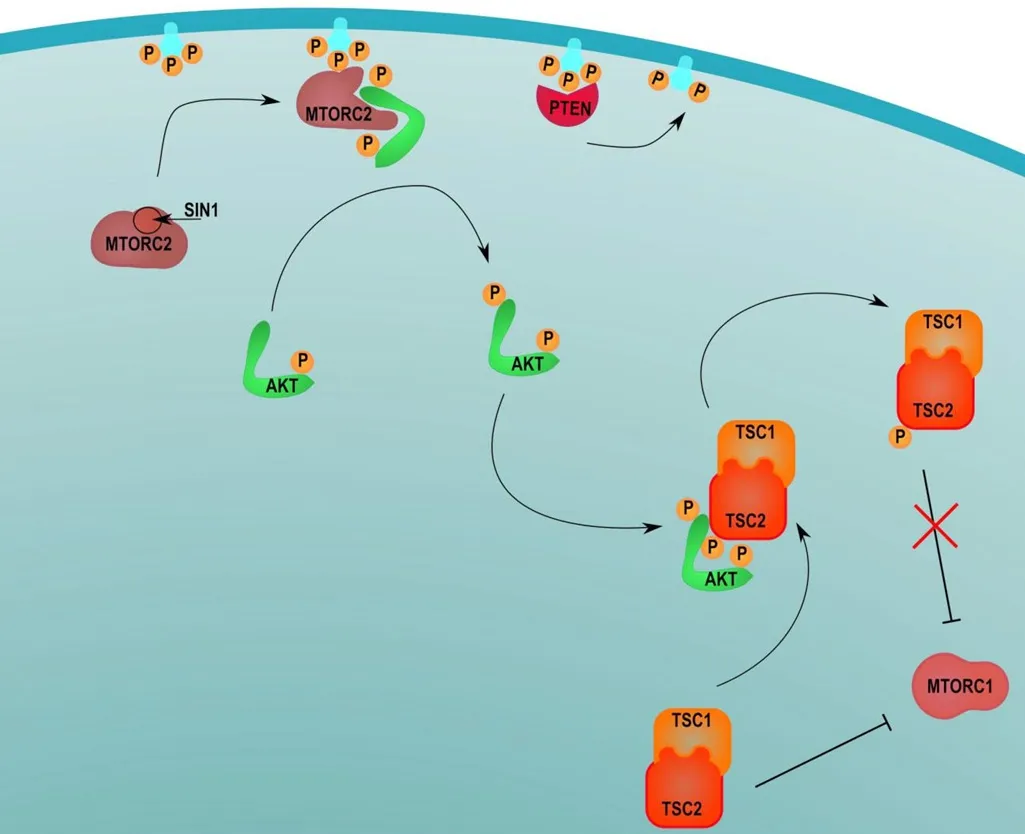
Figure 3.mTORC1 and two signaling pathways.This figure describes the transcriptional mTORC1 and mTORC2 pathways

Figure 4.Crosstalk with SHH pathway: this image describes the crosstalk between multiple pathways.SHH: sonic hedgehog; SMO:smoothened; TGF: transforming growth factor
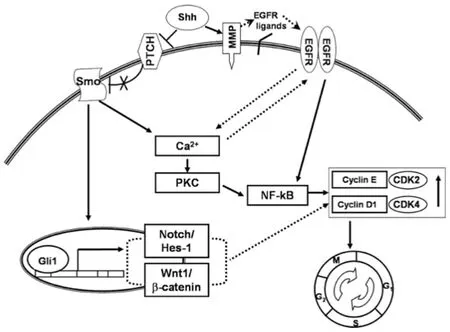
Figure 5.SHH pathway increases intracellular calcium, which can also increase the activation of EGFR.SHH also activates MMPs,which will activate the EGFR and increase cellular growth[71].SHH: sonic hedgehog; EGFR: epidermal growth factor receptor; MMP:metalloproteinases; NF-κB: nuclear factor-kappa B
Moreover,NOTCHgene mutation contributes to cSCC development through its role in chronic inflammation, trauma, or both.NOTCHgenes are responsible for the production of NOTCH proteins involved in cell proliferation and regulation of apoptosis[73].Cytochrome P450 family 1 subfamily B member 1(CYP1B1), a heme-thiolate monooxygenase, is involved in estrogen metabolism and biosynthesis as well as a catalyst to hydroxylation of E2 to 4-hydroxyestradiol.Normal keratinocytes have been found to express this enzyme.In fact, recent studies have found that CYP1B1 antagonizes the signaling of NOTCH1, which,in turn, blocks keratinocyte proliferation and differentiation.Western blot and immunofluorescence display that there is increased involucrin, keratin 10, and ki67 (a proliferation marker) after downregulation and knock out of CYP1B1, demonstrating augmentation of keratinocyte proliferation and differentiation[74].More research is being performed on the effect of CYP1B1 and NOTCH1 within SCC cells.This innovative research can lead to a new creation of a pharmacologic agent that can treat lacSCC in the future[74].
OtherNOTCHpathway genes include the recombination signal binding protein for immunoglobulin J gene (RBPJ).This gene codes for RBP-Jκ also known as “CBF1, Suppressor of Hairless, Lag-1” (CSL)transcription factor within keratinocytes.Upper epithelial cells downregulate the production of CSL.However, pre-malignant and cSCCs demonstrate increased levels of CSL, leading to the excessive proliferation of normal epithelial cells underlying the development of actinic keratoses and cSCCs[75].
Role of microRNAs
MicroRNAs (miRNAs) play an important role in cell growth of cSCCs.However, the function of the many miRNAs varies.Within various signaling pathways, some miRNAs are upregulated, while others are downregulated, causing various responses.However, all these miRNAs increase cell growth of cSCC malignant lesions.For example, miR-21 works as an oncogene that targets transcription factor GRHL3, an important element in the PTEN pathway.When miR-21 is upregulated, the PTEN and PI3K/Akt/mTOR signaling pathways are overexpressed leading to increased cell growth[76].This is one of the many miRNAs that influence cell growth of cSCCs.In fact, some of the upregulated miRNAs determine the aggressiveness of the cSCC lesion.Because miRNAs influence both the PTEN and mTOR pathways, this information always shows potential of crosstalk between the BCC and cSCC pathways.
Apoptosis evasion
An important cause of DNA instability is solar UV.Both UVA and UVB radiation induce the most common types of cSCC.It is well-established that UVB radiation causes direct keratinocyte DNA damage leading to cSCC, but exposure to UVA radiation has also been shown to foster tumorigenesis through DNA damage resulting from photosensitizers within the body causing indirect production of ROS.In natural sunlight, the ratio of UVA:UVB varies depending on the day, season, and latitude[77].It is not well understood how exposure to variable ratios of UVA:UVB radiation affects progression through these oncogenic pathways.However, use of sunscreen that blocks UVB without blocking UVA, as well as chronic or acute UV exposure through window glass, can possibly increase the ratio.Foods containing furocoumarins (psoralens) also may increase the absorption of UVA and thus alter the UVA:UVB ratio[78].Moreover, patients should be precautioned that substances applied to the skin such as retinoids, tanning oils and products with methyl and benzyl nicotinate may have the opposite effect of sunscreen and enhance the cutaneous absorption of UV radiation[79].
Solar UV radiation can induce DNA mutations within the cyclobutene pyrimidine dimers and 6-4 photoproducts[80].These mutations can activate the ataxia telangiectasia and Rad3 (ATR) DNA repair system that, in turn, activate TP53, a tumor suppressor important for apoptosis.The BCC sections elaborate on the mechanisms of ATR.Mutation in either or both genes can lead to apoptotic escape and resultant overactive cell growth.
Mutation of cutaneous TP53 thus evokes an uncontrolled activation of the cell cycle allowing mutated cells to progress to the synthesis phase (S phase) of the cell cycle prematurely.Wildtype CDKN2A/p16 is responsible for generating cyclin dependent kinase (cdk) inhibitors INK4A and p14, both of which are TP53 degradation inhibitors.INK4A binds to cdk4 and cdk6, inhibiting cell cycle progression into the S phase.When the oncogene CDKN2A/p16 is mutated, INK4A is mutated, resulting in a lack of inhibition of cdk4 and cdk6 within the cell cycle.This mechanism resembles that of TP53 in that the cell cycle prematurely progresses into the S phase allowing unregulated DNA replication and advancement into the mitotic phase of the cell cycle.Recent studies have discovered with flow cytometry targeting ATR-activated cells can eradicate the number of cells with 6-4 photoproducts.Eliminating these cells can block solar UV radiation-induced damage from occurring and potentially cSCC tumorigenesis [Figure 6].

Figure 6.Cell cycle displaying TP53 inhibition of cell proliferation.G1 to S phase by CDKN2A blockade of cdk4 and cdk6[81]
Downregulation of certain miRNAs can affect apoptosis; specifically, the downregulation of miR-34a decreases the function of TP53 leading to avoidance of apoptosis.Select solar UV radiated keratinocytes appear to have the downregulation of miR-34a, leading to the proapoptotic effect of these tumor cells[76].
Keratoacanthomas (KA) are controversial lesions.These lesions are classified as cSCCs or lesions with cSCC potential that can spontaneously resolve.Nevertheless, taking the risk of waiting for a KA to spontaneously resolve can be detrimental, since not all lesions regress.Some KAs continue to grow rapidly; a subset may become aggressive and metastasize.There are many gene mutations that cause the transformation of signaling pathways that eventually lead to the appearance of these lesions.The most common type of genetic pathway that is mutated is the apoptotic pathway.The most noteworthy upregulated genes found through microarray analysis in KAs wereMALAT-1,S100A8, andEHF.These genes modulate caspases,Bax, and Bcl-2, leading to avoidance of apoptosis[82].Microarrays show thousands of 1449 genes that vary from those of cSCC, yet there are many similar gross and histological features between cSCC and KA.Regardless of this debate, KAs have a highly malignant potential similar to those of cSCC and should be treated as soon as they are discovered.
Immune response evasion
UVB radiation (and to a lesser extent, UVA radiation) modulates the immune response over time.Actinic keratosis (AK) is a pre-cancerous lesion that may evolve into cSCC.Whether AK is to be viewed as a dysplastic, precancerous lesion of keratinocytes or if they should rather be understood to be highly scattered, disorganized cSCCin situhas been contested in the realms of dermatopathology and histopathology[83].Further research will be required to elucidate the proper classification of AKs.For this discussion, AKs will be considered precancerous lesions.The progression from AK to cSCC is associated with UVA and UVB modulation of immune signaling pathways.Chronic UV radiation causes an increase in p53 associated inflammation affecting apoptosis of keratinocytes; however, only roughly10% of AKs progress to cSCC.In one school of thought, the fate of AK is dependent a stepwise progression through several stages, driven by complex immunologic mechanisms.In many cases, AKs may present asymptomatically with little or no inflammation.These lesions may be subclassified as asymptomatic actinic keratoses (AAK).Although initially asymptomatic, some AKs become inflamed and present symptomatically and thus may be classified as inflamed actinic keratoses (IAKs).
It is thought that some AAKs progress to IAKs as cells undergo molecular and immunologic changes that promote increased growth and inflammation.For example, Fas ligand (FasL) site desensitization may lead to the inhibition of CD8+T lymphocytes resulting in impaired CD8+-mediated apoptosis and thus promote lesion progression[76].Other AAKs may not undergo the changes necessary for progression and remain asymptomatic.It has also been shown that IAKs recruit immune cells, including CD3+, CD4+, CD8+T lymphocytes, and Langerhans cells, producing further inflammation[84].Should an IAK experience sufficient growth and keratinocyte damage, it may progress to cSCCin situor Bowen’s Disease.Bowen’s Disease is slow-growing and shows displays only partial-thickness atypia.With further inflammation, damage, and mutations of keratinocytes, Bowen’s disease can evolve into cSCC with full thickness atypia and potential to invade and metastasize.The role of inflammation in the progression from AAK to IAK to cSCC is also supported by a stepwise increase in the expression of Bcl-2.Overall, the UV damage of keratinocyte DNA leads to the progression of a pre-cancerous asymptomatic lesion into a malignancy.
Secondary to UV induced cSCC are those induced through keratinocyte signaling mutation induced by chronic inflammation.Injury to the skin causes a wound healing cascade to occur, activating keratinocyte proliferation and many of the cell signaling pathways that are involved in tumorigenesis.The EGFR pathway, the intrinsic pathway, and the extrinsic inflammatory pathways are all included in the cell signaling pathways causing tumorigenesis.Chronic inflammatory conditions, including lichen sclerosus,may activate the intrinsic pathways causing chronic inflammation, increased cell turn over, and possible tumorigenesis by activating wound healing and repair simultaneously[85].The extrinsic pathway includes the influence of environmental factors and viral causes of inflammation, including human papillomavirus(HPV)[85][Figure 7].The viral mechanisms of HPV will be discussed later in the verrucous carcinoma section.This pathway involves the release of a cascade of inflammatory cytokines and chemokines which promote the migration of lymphocytes to the injury site and subsequent repair.Chemokines can enhance cell turnover causing increased DNA replication, predisposing cells to genetic mutations that foster the formation of tumors such as cSCC.In addition, cutaneous unilateral linear porokeratosis, another chronic inflammatory disease, has high potential for malignant transformation due to its mutations in psoriasin,p16INK4a, and involucrin, similar to that of cSCC but at a lower level[87].However, when the AKs and cSCC lesions do appear, they produce chemokines that increase metastatic potential.

Figure 7.This image shows the extrinsic pathway insults including infection, stress, UV radiation, and carcinogens while the intrinsic pathway is due to oncogene activation[86].UV: ultraviolet radiation; JAK: Janus kinase
Chemically induced mutagenesis
Cell growth and apoptosis
Chemicals, including arsenic and pharmacologic agents, may be implicated in the development of cSCCs.Chronic exposure to high concentrations of arsenic in food and drinking water is a common cause of cSCC, especially cSCC on the palms.Arsenic levels in drinking water as low as 300 μg/L have been found to cause cSCC[88].Levels between 200-1000 μg/L have been identified in 2013 in the ground water in multiple communities in Massachusetts[89].There are many medications and vaccinations that include the use of arsenic as adjuvants, active and inactive ingredients depending on the medication or vaccine;however, the amount within these pharmacologic agents is not enough to evoke malignant transformation of keratinocytes.Nonetheless, there is a need for further research on the chronic exposure of small amounts of arsenic and possible malignancies.Arsenic-exposed hyperkeratotic epithelial cells express elevated levels of keratin-1, keratin-10, involucrin, and loricrin, biochemical mediators important to the proliferation of keratinocytes[88].These lesions can further progress to become Bowen’s disease and later cSCC with fullatypia.Arsenic has also been found to change the functionality of transcription factors and transcriptional co-activators that affect cell growth and the stromal environment in which keratinocytes reside.Genetic pathways of UV-induced cSCCs commonly expressTP53,HRAS, or other tumor suppressor gene mutations leading to evasion of apoptosis.However, when analyzing the signaling pathway modulation caused by arsenic, many of the genes mutated are directly involved with transcription.Nrf-2, a transcription factor important to the homeostasis of redox reactions within keratinocytes, keeps inflammation and oxidative stress at bay.Gene suppression of Nrf-2 in chronically exposed arsenic human epithelial cell lines produces arsenic-induced malignant conversion of keratinocytes[88].Additionally, the transcription co-activator,Yap, effects keratinocytes leading to malignant transformation.Yap is only activated via phosphorylation by Phospho-LATS kinase in wild-type keratinocytes.With a high level of chronic arsenic exposure, Yap is translocated to the nucleus of keratinocytes causing increased proliferation and the eventual appearance of dysplasia and malignancy[90].
BRAF inhibitors can also affect the appearance of cSCCs.During treatment with BRAF inhibitors, cSCCs can spontaneously appear.There are many case reports showing that BRAF inhibitor, Sorafenib, inhibits PI3K, MAP kinase, and NFκB, which can reduce the release of cytokines from Langerhans cells, decreasing T lymphocyte response to new cSCCs[91].As a result, the presence of IAKs or small cSCCs on patients taking BRAF inhibitors has the potential to cause rapid growth of cSCCs to become lacSCC due to the lack of immune response against these lesions.
Supporting stroma and vascularization
Invasion of the stroma that supports keratinocytes is a process that maintains and even evokes aggressive malignant behavior in cSCC.Arsenic and UV radiation affect keratinocyte stroma similarly.Chronicarsenic exposure leads to hypersecretion of MMPs, which degrade type IV collagen in the basement membrane.This promotes the diapedesis of immune cells and the release of inflammatory markers and chemokines, causing further cell growth and increased malignant potential.In addition to arsenic,UV also affects the stroma of underlying keratinocytes.UV radiation leads to genetic changes within the cell affecting dermal fibroblasts and the underlying basement membranes.The structure of the microenvironment, including the epidermal basement membrane and dermal extracellular matrix, can be altered by precancerous lesions.These precancerous lesions cause a release of chemokines, leading to an invasion of inflammatory cells and encourage the growth of cSCCs.These AKs and cSCCs have been found to have increases in MMPs, disintegrin-like metalloproteinases domain (ADAMs), tissue inhibitors of metalloproteinases, and other extracellular matrix degrading enzymes.Furthermore, tumor cells change the molecular function of cell-cell adhesion and prevent the integrins of damaged basement membrane from binding to extracellular matrix and binding to tumor cells.Tumor cells upregulate complement factor H and factor H-like protein 1, leading to further insult and invasion.The upregulation of these inflammatory signals by tumor cells directly affects the microenvironments of keratinocytes, causing further insult to the stroma[92].
Trauma and chronic inflammatory conditions
Chronic inflammation and trauma can change the epigenetics of the composition of the underlying stroma on which the epithelial cells lie, allowing these cells to have greater metastatic potential.The upregulation ofSTAT3,p63,FGFR2, and other genes coding for and upregulating chemokines can be the change necessary for conferring invasive potential[92].The chemokine SDF-1 and its endothelial cell receptor C-X-C chemokine receptor type 4 (CXCR4), both involved in angiogenesis, are upregulated in chronic inflammatory states.Once premalignant lesions are formed, SDF-1 and CXCR4 may affect chemotaxis,upregulation of metalloproteins, and activation of stromal fibroblasts, leading to loss of collagen types in the basement membrane[93].Marjolijn ulcers (seen in burn patients), chronic decubitus ulcers, and diabetic ulcers can all undergo processes that lead to stromal changes and subsequent keratinocyte differentiation.The signaling pathways associated with chronic inflammatory ulcer transformation can appear from a spontaneously new pathway that is also associated with the epithelial cell growth.One signaling pathway is linked with inhibiting the cdk, PDGF, and SHH pathway modulations.However, an additional pathway deals with the suppression of the proapoptotic WNT/β catenin pathway.Both pathways downregulate the extracellular matrix genes and upregulate MMP gene activation, affecting many of the same pathways affected by UV radiation.Nevertheless, Marjolijn ulcers trigger a fibrotic change in the basement membrane causing a transformation in the function of adhesion molecules between keratinocytes[94].Marjolijn ulcers display decreased expression of IL-18, suggesting that overexpression and utilization of the immune system in chronic inflammation can eventually cause suppression of the immune response towards infections and cancerous cells[83].This information can guide further research into chronic inflammatory conditions and trauma causing cutaneous malignancies.
Not all cSCCs are preceded by precancerous lesions.Some cSCCs can appearde novo.This is especially true in the cSCCs developed by transplant patients.Transplant patients often have weakened immune systems due to both immunosuppressive therapy and the underlying conditions that necessitated the transplant.Post-transplant patients with cSCC have been found to havede novoT lymphocyte mutations inZNF577coding for zinc finger proteins andFLOTgene coding for T-cell migration, dampening the immune response against tumor cells and subsequent overgrowth[95].These types of genetic mutation pathways that lead to cSCC can be investigated as innovative treatment approaches for patients with more advanced forms of cSCC.
The tumorigenesis of cSCC is complex.UV exposure, exposure to carcinogenic substances, alteration of the stromal environment of keratinocytes, and chronic inflammatory states may all be implicated inthe development of a tumor, either independently or in combination.There are multiple possible genetic drivers capable of fostering the development of cSCC.Each of these drivers may influence the development of a malignancy by impacting one of the four hallmark pathophysiological processes common to all cancers.Depending on the provoking carcinogenesis, signaling mutations of the provoking carcinogenesis,and crosstalk between the signaling of the hallmark processes, cSCC may have various aggressive potentials and require various biogenic signaling-related therapies.This is a complex multi-factorial individualized process overlapping signaling mutations that can be individually addressed therapeutically for cSCC.It is important to note that all tumor cells are not created equally.In fact, the processes, mutations, and pathways discussed above can occur at once within one tumor between multiple cells.GEP testing by Castle Biosciences, Phoenix AZ, is being developed for SCC to assess gene upregulation and downregulation within a given tumor for high and low risk patterns.This test will help determine metastasis risk among high risk cSCC patients.Thus, the GEP test, when available, should help improve prognosis projections for thus tumors labelled as poor prognosis cSCC[96].
GENETICS OF VERRUCOUS CARCINOMA
Verrucous carcinoma (VC), first described by Dr.Lauren Ackerman in 1948, is a rare variant of SCC,usually in the oral mucosa.Risk factors for VC include HPV infection, smoking, chronic inflammation,and repeated trauma.While the tumor generally has a good survival prognosis, it can be locally aggressive and may recur after treatment.The mortality with VC is more often due to local invasion rather than metastases[97].VC usually presents as a warty, exophytic, non-ulcerating lesion with a red/white surface,making it difficult to clinically distinguish from other similar appearing dermatologic conditions.This ambiguity leads to a lack of objective criteria for diagnosis, as well as challenges in establishing a definitive method of treatment.
Malignant cell growth
While a definitive association has not yet been described, HPV infection may be a significant contributor to VC.Plantar VC was shown to be associated with HPV type 16, while VC of the scalp was associated with HPV type 33[98,99].E6 and E7 oncoprotein production by high-risk strains of HPV cause p53 and retinoblastoma (RB) tumor suppressor inactivation, leading to cell cycle disruption and contributing to the progression of VC[100].The E6 protein forms a trimeric complex with p53, a tumor suppressor, and E6-AP, a cellular ubiquitination enzyme [Figure 8].This complex interferes with the functions of p53, leading to uncontrolled cell growth.The RB protein normally binds to transcription factors of the E2F family,which allows it to suppress replication enzyme genes like origin recognition complex 1, mini-chromosome maintenance proteins, and cell division cycle 6[101].The E7 protein inhibits this interaction between E2F and RB, allowing for uninhibited cell division and replication[102].

Figure 8.The pathway of human papilloma virus (HPV).This figure describes the effects of HPV on cells involving the E7 and E7 proteins
Chronic inflammation is another hypothesized cause of cutaneous VC.VC has arisen in cases of decubitus ulcers, hidradenitis suppurativa, epidermolysis bullosa, as well as chronic bladder inflammation by schistosomiasis[103,104].Chronic inflammation causes oxidative stress due to the release of cytokines,prostaglandins, and TNF.This leads to genetic and epigenetic modifications including the inhibition of DNA repair, modification of transcription, prevention of apoptosis, and stimulation of angiogenesis.Inflammatory cells such as macrophages and T lymphocytes may express migration inhibitory factor (MIF),which inhibits p53 transcriptional activity, leading to a loss of cell cycle regulation.The Cys81residue of MIF is thought to play a critical role in this association between MIF and p53, leading to the inhibition of p53 mediated gene activation and apoptosis[105].The loss of p53 contributes to a lack of response to DNA damage, which increases the likelihood of carcinogenesis.
Repeated mechanical irritation also leads to DNA damage and resultant carcinogenesis[103].Specifically, in the case of epidermolysis bullosa, VC was thought to arise due to a mutation in the keratin 5 gene (KRT5).In keratinocytes, KRT5 and KRT14 normally copolymerize to form intermediate filaments which help anchor the epidermis[106].Mutations of helix boundary motifs of KRT5 are thought to lead to the most severe phenotype of epidermolysis bullosa[107].These mutations lead to fragile keratinocytes with unstable cell-to-cell junctions.This instability causes keratinocyte-mediated inflammation, which may contribute to the pathogenesis of VC[104].
Oral verrucous carcinoma (OVC) is more commonly seen than the cutaneous subtype.The PI3K/AKT/mTOR pathway was shown to be upregulated in OVC, suggesting a role for this pathway in the progression of OVC[59].In this pathway, PI3K phosphorylates AKT, which confines AKT to the plasma membrane[Figure 2].This is followed by many downstream effects of AKT, one of which is the activation of mTOR.mTOR is a serine/threonine kinase expressed in human cells, and its action is executed by two complexes,TORC1 and TORC2.TORC1 leads to the downstream activation of 4EBP1 and P70S6K, which are involved in the translation of mRNA into proteins for cell growth[108].This pathway leads to increased proliferation of tumor cells, as well as resistance to treatment.The PI3K/Akt/mTOR pathway is also upregulated in oral SCC.
Furthermore, the downregulation of miRNAs contributes to the pathogenesis of OVC.Specifically, miR-195 is found to be significantly downregulated in OVC[109].miR-195 serves as either an oncogene or a tumorsuppressor, and is involved in tumor proliferation, metastasis, apoptosis, and angiogenesis.miR-195 exerts its effects through various target genes, one of which is cdk6.cdk6 is activated in the G1 phase of the cellcycle and plays a role in the development of various cancers; cdk6 is found to be significantly upregulated in OVC[110].This negative correlation between the expression of cdk6 and miR-195 in OVC serves as confirmation of the interaction between the two.However, the specific role of miR-195 and cdk6 in OVC pathogenesis is an area requiring further investigation.
Apoptosis evasion
The diagnostic value of expression of the apoptosis-related regulatory proteins p16, p53, p21, and RBGP(retinoblastoma gene product) in cases of oral, penile, and cutaneous VC has been studied.The variation in expression of these proteins may be helpful in differentiating VC from classic SCC[111].
P16 is a cyclin D-dependent kinase inhibitor that is inactivated in a variety of cancers.This protein blocks the progression of the cell from the G1to S phase[112].Interestingly, p16 additionally plays a role in the regulation of UV-induced apoptosis of cells[113].In cases of VC, some level of p16 expression is found in 79%of cases[111].p21, a mediator of p53 function, is upregulated by p53 in response to DNA damage.Similar to the actions of p16, p21 upregulation leads to the inhibition of cdk2 and CD4 complexes, causing cell cycle arrest in the G1phase[111].In 58% of VC cases, p21 expression is seen in the lower third of the epithelium,while the remaining 42% of cases show full-thickness expression[111].
While p53 mutations are one of the most frequently reported tumor suppressor mutations in human cancers, p53 expression is significantly different in VCvs.SCC.All cases of SCC showed p53 expression throughout the entire thickness of the epithelium, yet cases of VC showed p53 expression exclusively in the lower third of the epithelium[111].Interestingly, the Ki67 protein showed a very similar pattern of expression in VCvs.SCC, with the former showing expression only in the lower third of the epithelium.The exact function of Ki67 is yet to be understood; it is thought to play a role in regulation of the G1, S, G2, and M phases of the cell cycle[114].Both p53 and Ki67 showed a statistically significant difference in expression between VC and SCC and may play a role in differentiating these tumors.
Supporting stroma and vascularization
The differences in angiogenesis between SCC and VC have been studied using measures such as microvascular density (MVD) and endothelial proliferative index (EPI).MVD is measured by scanning CD34 immunostained sections to find areas with a high density of microvascularization.EPI is estimated using MIB-1 stained slides to locate MIB-1 positive endothelial cells.Both MVD and EPI were found to be significantly lower in VCvs.SCC; it is hypothesized that the less aggressive nature of VC may be attributable to these lower values[115].
The release of chemokines due to chronic inflammation also plays a role in angiogenesis.Members of the CXC chemokine family that are glutamate-leucine-arginine motif positive (ELR+) exert their proangiogenic effects by activating the CXCR2 receptor.This activation leads to increased levels of VEGF and decreased levels of thrombospondin-1, an antiangiogenic glycoprotein[116].Stromal cell derived factor known as SCDF1 or CXCL12 plays a role in increasing endothelial VEGF expression, as well as the chemotaxis of cancer cells[117].
Modulating immunity
Chronic inflammation correlates with a lack of tumor immune surveillance.The cytokine IL-23 is upregulated in many human tumors.While IL-23 increases angiogenesis and upregulates matrix metalloproteases, one of its functions is to inhibit CD8+T-cells, which are an essential component of tumor surveillance.Deletion of IL-23 was shown to restrict tumor growth, leading to protection against carcinogenesis[118].
The use of a TNF inhibitor, specifically etanercept, is believed to cause immune modification that may ultimately lead to the development of cutaneous VC in a patient[119].Patients undergoing TNF-alpha inhibitor therapy may be susceptible to complex effects on the regulation of inflammation, autoimmunity,and apoptosis.These drugs have the potential to inhibit the extrinsic apoptotic pathway by modulating the activation of death receptors 4 and 5 (DR4 and DR5) [Figure 7].DR4 and DR5 are normally activated by TRAIL, resulting in the apoptosis of cancer cells[119].Drugs like etanercept inhibit this process, potentiating carcinogenesis.Nonetheless, a systematic review and metanalysis of patients treated with anti-TNF-α agents failed to demonstrate any increased risk of malignancy[120].
Furthermore, there is a possible effect of anti-TNF therapy on hypermethylation of the ASC/TMS1 protein.ASC/TMS1 is responsible for the activation of phagocytes through the secretion of cytokines and activation of the inflammasome[119].ASC/TMS1 also indirectly potentiates apoptosis through its interaction with p53 and the Bcl-2-associated X protein (BAX).BAX induces apoptosis, due to the activation of caspase 8.ASC/TMS1 may be responsible for translocation of BAX to mitochondria by serving as its carrier protein[119].Hypermethylation of ASC/TMS1 inhibits these processes, enhancing cancer development.The effect of anti-TNF drugs on ASC/TMS1 function still requires further investigation.With these known genetic signaling pathways, pharmacogenomic treatment plans can be focused on the patient and/or lesions specific genetic pathways in conjunction with the anti-TNF drugs to target all mutated signaling that can appear within patients’ varying flux states.
GENETICS OF BASOSQUAMOUS CELL CARCINOMA
Basosquamous cell carcinoma (BSC) is a rare, aggressive skin neoplasm that has histopathological characteristics of both BCC and SCC[121].The majority of BSC cases are found in the head and neck region,with older Caucasian males most commonly affected[122].Clinically, BSC is indistinguishable from BCC and is commonly referred to as “metatypical basal cell carcinoma[122,123]”.Diagnosis of BSC is typically made with a biopsy of a lesion suspected of being a BCC or an SCC[124].It should be noted that BSC is distinct from a collision tumor in that BSC is more of a “mixed tumor” consisting of BCC with areas of SCC differentiation[125], whereas a collision tumor has distinct BCC and SCC entities that are present in close proximity[126].This can be seen histologically by the presence of a transition zone of atypia in BSC, an uncommon feature for a collision tumor[125].Compared to BCC, BSC has a higher tendency for local recurrence and a higher propensity for lymph node and distant metastases[121,127].BSC comprises approximately 2% of all skin cancers with a metastasis rate of about 7%[128]compared with a metastasis rate of BCC of 0.0028%-0.55%[127,129]and of SCC of 2%-5%[130].The BSC recurrence rate is 12%-51% after standard surgical excision[128].This contrasts with post-standard surgical excision recurrence rates of BCC and SCC at 5%-14%[131]and ~8%,[132,133]respectively.BSC has an approximately 4% recurrence rate following Mohs micrographic surgery compared with recurrence rates post-Mohs micrographic surgery (MMS) of BCC (1%-2%)[131]and SCC (3%), respectively, although there was some variability in the literature[128,131,134].Interestingly, the recurrence rates of BSC post-MMS were reported to be about 4% regardless of site (head and neck, trunk, lower limb, or upper limb).In BSC larger than 2 cm, the rate of recurrence is believed to be slightly increased[128]; however, these results were not statistically significant, likely due to small sample size[134].Thus, MMS is currently the preferred treatment for BSC independent of body location.
Malignant cell growth
BCC genetic drivers’ role in BSC
BSCs are frequently associated with SHH pathway mutations, implicating SHH deregulation as the primary driver in BSC and providing evidence that BSCs shares similar cancer drivers to BCC[135].Loss of function inPTCH1and gain of function in G-protein-coupled receptorSMOin the BCC pathway are the most common mutations that cause SHH deregulation in the BCC pathway.Similarly, 45% of BSCs were shown to have deleterious mutations inPTCH1compared to 44% of BCCs and 10% of SCCs.About 5% of BSCscontainSMOoncogenic mutations compared with 25% of BCCs and 0% of SCCs[135].BSCs also contained all the other known BCC cancer drivers,MYCN, PP6C, GRIN2A, CSMD3, DCC, PREX2, APC, and ARID1A at a frequency statistically similar to that of BCCs[135].
SCC genetic drivers’ role in BSC
BSCs lack the classical SCC driver mutations,NOTCH1/2, HRAS/KRAS,andCDK2NA,at a frequency expressed by SCCs.Instead, BSC contain SCC driver mutations at a significantly lower rate that is closer to that seen in BCCs.The presence of classical BCC cancer drivers in BSCs and the lack of classical SCC driver mutations suggests that BSCs have a mutational landscape similar to that of BCC, and that BSC cancer drivers likely arise through the deregulation of SHH signaling[135].
Genetics of cells modulating between BCC to SCC cells
It is believed that BSC originally derive from BCC due to their similar mutational landscape; they subsequently acquire mutations that lead to squamatization[121].Of the 20 cancer genes identified in BSC,ARID1Ais mutated in 45% of BSCs compared to 19% of BCC and 19% of SCC[125].ARID1Anormally plays a role in the differentiation of several cancer types because it encodes a component of the chromatin remodeling complex, SWI/SNF.SWI/SNF plays an important role in repairing damaged DNA, soARID1Aimpairment can cause defects in DNA repair.Disruption ofARID1Areduces the restrictive nature of chromatin remodeling in terminally differentiated cells, imparting a plasticity that increases cell survival,regeneration, and proliferation.Under selective pressure, such as a SMO inhibitor treatment,ARID1Amutations allow keratinocytes to undergo squamatization, promotingde novoSCC development from BCC[125].Further research could be done to see if mutations within other components of chromatin remodeling could confer plasticity in BSC.
Additionally, switching from the SHH pathway to the RAS/MAPK pathway is a form of crosstalk between the pathways and another way that BCC can avoid selective pressures such as SMO inhibitors,leading to squamatization.As discussed in the SCC section, an upregulated RAS/MAPK pathway can lead to SCC.Pathway switching is believed to be driven by a loss of primary cilia, a microtubule-based signaling organelle that is essential for high SHH pathway signaling, promoting RAS/MAPK signaling and subsequent squamatization as seen in Figure 9.Furthermore, Gli1 is active in the nucleus and is typically elevated in BCC.Stained BSC shows that basaloid keratinocytes demonstrate high Gli1 and low MAPK staining which is similar to that of BCC.Squamatized keratinocytes in BSC demonstrate low Gli1 and high MAPK staining which is similar to that of SCC.Interestingly, the transition zone shows a moderate MAPK expression and high Gli1 expression, indicating that RAS-MAPK pathway activation drive squamatization in BSC with subsequent loss of Gli1 expression as a secondary event.This discovery indicates the activation of the RAS-MAPK pathway and subsequent reduction in SHH signaling as a potential modulator of tumor plasticity in BSC[136].Overall, tumor fate in BSC is believed to be dictated by a balance between SHH and RAS/MAPK signaling[125].

Figure 9.Genetic lineage of BSC.A: activating SHH pathway mutations initially drive formation of BCCs.Acquisition of de novo ARID1A mutations or other chromatin remodeling mutations under pharmacological SMO inhibition drive cellular plasticity, pushing basal cells to undergo squamatization and leading to BSC formation; B: within the BCC zone, high levels of SHH signaling, low RAS/MAPK pathway activity, and high levels of ciliation drive tumor growth.Within the SCC zone, RAS/MAPK signaling increases with a concomitant reduction in SHH pathway activity and ciliation.Within the transition zone, ce3lls begin to show higher levels of RAS/MAPK pathway activity while maintaining SHH signaling.The levels of ciliation are unknown.SHH: sonic hedgehog; BCC: basal cell carcinoma; SMO:Smoothened; BSC: basosquamous cell carcinoma
Calcium flux is a potential modulator of the balance between SHH and RAS/MAPK signaling in BSC.The degree of extracellular calcium is a regulator of the SHH pathway.In rat gastric mucosal cells, SHH is unable to activate extracellular signal-related kinases in calcium-free conditions.Conversely, cells in a calcium-rich media show increased intracellular calcium levels, a marker for SHH pathway activation[137].Altogether, it is possible that calcium flux shifts the balance between SHH and RAS/MAPK signaling in BSC, contributing to tumor plasticity[138].More research is needed to establish a causal relationship between calcium flux and the balance of SHH and RAS/MAPK.Future studies could be done to identify additional modulators and to understand the interplay between existing modulators in conferring tumor plasticity.
Though it is known that BSC appear from BCC, it is unknown if these cutaneous lesions can also appear from a SCC.However, due to the consistent state of flux that the body undergoes and crosstalk betweenthese genetic pathways, there is a possibility that SCCs could become BCCs and BCCs can become SCCs.There is a need for further research finding data to address this issue.
Prevention of apoptosis
Strong expression of FasL in both BCC and SCC is believed to allow the tumor to avoid apoptosis.FasL mediates apoptosis in cells that express Fas receptor and is a member of the tumor necrosis family.FasL on tumor cells can bind to the Fas receptor on T-cells, macrophages, and natural killer cells which make these effector cells unable to infiltrate the tumor nodules and trigger the extrinsic apoptotic pathway[139].Although there have not been studies confirming that strong expression of FasL is an important mode for how BSCs prevent apoptosis, BSCs’ genetic similarity to BCC and histopathologic characteristics of bothBCC and SCC suggest that strong FasL expression plays a role in allowing BSC to avoid apoptosis.BSC that do not have strong expression of FasL may not be able to avoid apoptosis or may avoid apoptosis through other means.
Additionally, bcl-2 promotes cell survival without stimulating cell proliferation.Bcl-2 is associated with the intrinsic apoptotic pathway, triggered by intracellular stress[140].Studies indicate that bcl-2 positive tumors have a better prognosis and slower course of progression.In BSC, immunohistochemical studies for bcl-2 yield a positive result in the BCC regions but a negative result in both the transition zone and SCC region[141].Studies show only low-grade bcl-2 positivity in the BCC regions of BSC, similar to that of aggressive BCC.In contrast, non-aggressive BCC such as nodular and superficial BCC have high grade bcl-2 positivity.This suggests that BSC may downregulate bcl-2 in the BCC regions, stimulating uncontrolled cell proliferation and contributing to its more aggressive behavior compared to non-aggressive BCC[141].
Crosstalk
Crosstalk between various signaling pathways can be a determining factor as to why some patients respond to cancer treatments and others fail treatments.In fact, the BSC pathways and hair follicle pathways appear to have significant crosstalk; both of these pathways are dependent on SHH, PTCH1, SMO, and Gli[142].In normal hair follicle development, PTCH1acts as a receptor for SHH, and their interaction triggers SMO which modulates a signaling cascade.Activation of the SMOcascade results in subsequent translocation of Glito the nucleus.Gliis a key modulator of the SHHpathway and causes upregulation of several downstream target genes that modulate hair follicle development[143].Injury to skin enlists hair follicle stem cells in the healing process, namely through the SHHpathway.However, if cells that carry a mutation inSMOare recruited to the injury site, this mutation will trigger unregulated downstream SHHpathway signaling which is implicated in BCC and BSC[144].This shared SHHpathway involvement suggests that the mechanisms of tumor formation and hair follicle proliferation are interlinked and essential for the normal development of hair follicles with dysregulation or mutation in this pathway leading to cancer[143].It is possible that BSC originates as BCC from pluripotent basal cells and initiates in a similar manner as BCC, typically from hair follicle stem cells in the hair follicle bulge[145], as seen in the previous BCC section.Through modulating pathways conferring plasticity as described above, squamatization occurs to form BSC.
Vismodegib is a SHHpathway inhibitor that is used for the treatment of BCC.There have been many cases of SCC developing from BCC following treatment with Vismodegib.The appearance of SCC may indicate ade novoSCC adjacent to the BCC which is proliferating independently, an SCC that develops as a result of stem cell differentiation during SHH inhibition, or an SCC that was present as part of a metatypical BCC such as BSC that has decreased SHH signaling due to SHH inhibition and subsequently increased RAS/MAPK signaling[146].There fails to be many reported cases of SCC transforming to BCC following use of Vismodegib.Thus, it may be difficult to determine if SHH inhibitors could play a role in the desquamatization of cSCC to BCC.Nevertheless, because BSC has a high tendency for recurrence and has a high rate of metastasis, Moh’s micrographic surgery is currently the preferred treatment for BSC[128].
Supporting stroma and vascularization
When comparing supporting stroma of BCC and BSC, there is a significant difference between the stroma of BSC, and high risk (HR) BCCs (micronodular and/or infiltrative) compared to that of low risk (LR)BCCs (nodular and/or superficial).LR BCC group lesions have fibromyxoid stroma without desmoplastic stroma, whereas 80% of the HR BCC and 61.8% of the BSC group lesions have desmoplastic stroma[147].
The stromal differences in LR and HR BCCs and BSC further suggests variety between the tumor types.Fibromyxoid stroma is most often observed in lesions with mild inflammation.However, desmoplasticstroma is associated with dense inflammation which attributes to stroma-derived factors that recruit additional inflammatory cells or inflammatory cell secreting factors that change stromal characteristics[147].Inflammatory cell/secreting factor recruitment of collagenolytic enzymes that cause destruction of the surrounding collagen and matrix metalloproteinases that cause proteolysis of extracellular matrix components are implicated in SCC and likely BSC to cause desmoplastic stroma, facilitating tumor growth and metastasis[148].Further research needs to be done to identify the types and functions of immune cells in peritumoral inflammation in order to understand the interactions between peritumoral inflammation and stroma specifically in BSC.Additionally, activated fibroblasts and inflammatory cells of peritumoral stroma secrete extracellular matrix proteins and growth factors in a paracrine fashion which can change the expression of genes affecting angiogenesis, tumor growth, and metastasis.Though the precise mechanisms underlying the complex interactions between the stroma and tumor in BSC is unknown, the crosstalk communication between various genetic signaling pathways indicates the flux state amongst signaling pathways and keratinocyte homeostasis.
Modulating immune response
Dense peritumoral/perivascular inflammation in BSC is a marker of host immune response and may be associated with recruitment of protumor immune cells.For LR BCC, 83.3% of the lesions exhibit only mild peritumoral inflammation whereas of the HR BCC, 76% of the lesions exhibit dense peritumoral inflammation.Of the BSCs, 51.4% demonstrate dense peritumoral inflammation and 37.1% demonstrate moderate peritumoral inflammation, frequencies that most closely approximated those of the HR BCC group[147].Similarly, perivascular inflammation in adjacent dermis was observed in 91.4% of BSC group lesions, 97.5% of HR BCC group lesions, and 55% of LR BSC group lesions.Only the BSC and HR BCC group lesions were found to have dense perivascular inflammation[147].
The host immune response between BSC and HR BCC shows some similarities based on peritumoral and perivascular inflammation density, both in contrast to LR BCC.Firstly, differences in inflammation density between BSC and LR BCC may be attributed to an increased number of regulatory T-cells in BSC that suppress anti-tumor T-cell response and are associated with a worse prognosis compared to LR BCC that do not have increased regulatory T-cells[147].Secondly, immature dendritic cells (IDCs) are involved in attenuating the immune response in BCC and perhaps also in BSC.Attenuation of the immune response occurs through the induction of peripheral T-cell tolerance by IDCs and through regulatory DC secretion of IL-10 that suppresses T-cell proliferation.IDCs are also believed to contribute directly to tumor proliferation through unknown mechanisms.Lastly, an increase in expression of Th2 cytokines, specifically IL-4 and IL-10 contribute to an immunosuppressive tumor environment that favors proliferation in BCC and perhaps also BSC[149].It should be noted that the tumor permissive mechanisms described above likely occur concurrently with immune responses directed towards tumor eradication.Anti-tumor immune responses include: CD8+T-cells for a specific adaptive antitumor response, IL-23 for enhanced proliferation of memory T-cells, IL-12 for activation of mature DCs and Th1 antitumor immunity[149].Overall, BSC modulation of immune response appears to occur dynamically with a protumor, attenuated immune state only partially compensated for by the host antitumor response.This dynamic state shows myriad fluctuations and crosstalk signaling between genetic signaling pathways, which can further personalized BSC treatment for advanced or metastatic BSCs.
GENETICS OF MELANOMA
Although NMSC are the most common type of skin cancer, cutaneous melanoma is the most aggressive,accounting for greater than 80% of skin cancer deaths largely due to its susceptibility to metastasize to other organs[150].Close to 200,000 cases of melanoma will be diagnosed in the United States in 2020 with a rate of one American dying from melanoma every hour[151].There are 4 main types of melanoma: superficial spreading melanoma (SSM), nodular melanoma (NM), lentigo maligna melanoma (LMM), and acrallentiginous melanoma (ALM)[152].Cutaneous SSMs account for 70% of melanomas, while NMs account for 15%-30% of melanomas.Both SSM and NM subtypes are often associated with theBRAFV600E mutation[152].ALM (2%-10%) and LMM (5%) are rarer types that are both associated with mutations in the C-kit gene[152].
The transformation of melanocytes into melanoma cells and further progression to metastasis involves the complex interaction of signaling pathways with multiple environmental, genetic, and host factors.The primary contributor is DNA damage from UV light exposure, although genetic disorders like xeroderma pigmentosum and familial history of melanoma can strongly increase a person’s risk[153].Occupational exposure to ionizing radiation among radiologic technologists has been shown to increase risk to developing skin cancer and melanoma[154].A recent literature review suggests airline pilots and cabin crew may have twice the risk of melanoma compared to the general population and increased melanoma mortality among pilots from possible cumulative cosmic radiation[155].However, much of the relevant evidence is considered out-of-date as it reflects environment and behaviors in the late twentieth century and is incongruent with modern day standards.Immunosuppressed populations like organ-transplant recipients have an increased risk of many malignant cancers, with skin cancer being the most common[156].Understanding the mechanisms in various signaling pathways can provide insight into providing personalized and effective treatment.This amalgamation of genetic crosstalk modulates the transformation process and involves the hallmarks of cancers: (1) cell growth; (2) prevention of apoptosis; (3) supporting stroma and vascularization; and (4) modulating immune response.
Cell growth
UV-induced damage
Within hours, UVA causes immediate and lasting hyperpigmentation (tanning) due to induction of oxidative stress in melanocytes.UVA damages the extracellular matrix and induces an immune response facilitating invasion and metastasis of skin cancer cells[113].In contrast, UVB directly causes skin cancer genesis and induces a slower delayed tanning through a nascent melanin synthesis pathway and melanocortin receptor-1 (MC1R) signaling[3].MC1R activates the DNA damage response causing the formation of cyclobutene pyrimidine dimers and 6-4 photoproducts that distort the DNA helix and when unrepaired lead to mutations[157].In addition, UVB triggers an inflammatory response by recruiting neutrophils and macrophages and promotion of angiogenesis contributing to melanoma cell survival and metastasis[3].
CDKN2A/p16 deletion
The CDKN2A/p16 protein is a crucial cell cycle gatekeeper at the G1-S checkpoint and its location encodes for tumor suppressors p16INK4aand p14ARF[158].As mentioned in the cSCC section, P16INK4ainhibits cdk4 and cdk6 [Figure 10], activating RB protein and preventing cell cycle progression into S from G1 phase[159,160].On the other hand, p14ARFpositively regulates p53 [Figure 10] by inhibiting negative regulator MDM2[161,162].Deletions in the CDKN2A locus was found in 50% of all melanomas and had high penetrance in familial melanoma[153,163].The inactivation of CDKN2A and p53 inactivation leads to uncontrolled cell proliferation of melanocytes.
Apoptosis evasion
BRAF in RAS/RAF/MAPK/ERK pathway
The most prevalent and highly studied oncogenic melanoma mutation isBRAFwith the most common mutation substituting glutamic acid for valine (V600E)[164].An estimated 40%-50% of mutated melanomas are ofBRAFV600E mutation[165].In the MAPK pathway [Figure 10], the presence of the oncogenicBRAFV600E mutation drives constitutive phosphorylation of MEK1 and 2, activating ERK 1 and 2 and reprograming cellular metabolism to sustain cell survival and growth[166].In major melanoma subtypesof NRAS, BRAF, and c-kit pathways, proteins expressed in the MAPK signaling pathway has been found suggesting crosstalk between pathways[167].In addition, several mutations of key upstream proteins of the aforementioned MAPK cascade includes NRAS, KIT, GNAQ, and GNA11[168].In over 80% of uveal melanomas, GNAQ or GNA11 is activated using the MAPK pathway[169].
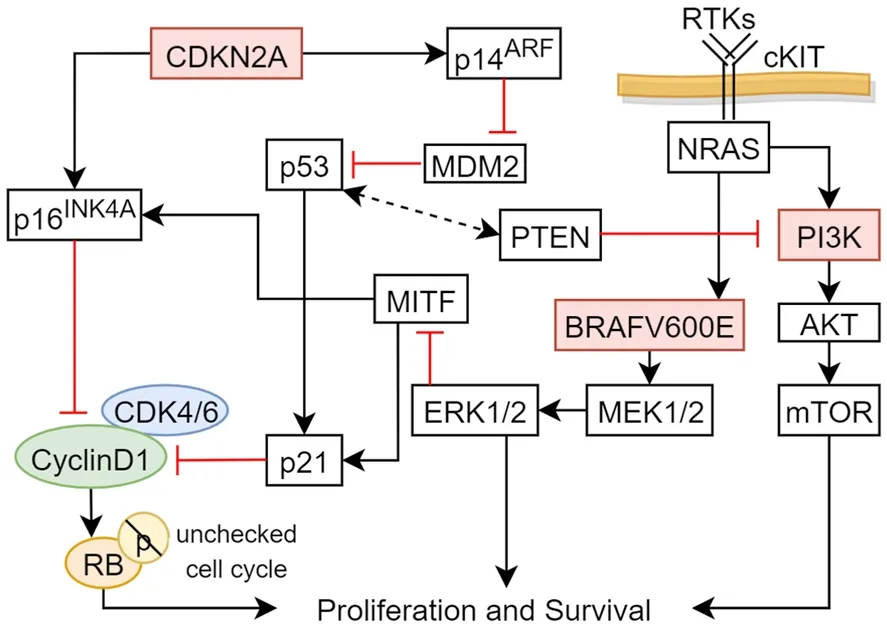
Figure 10.Significant signaling pathways in melanoma.Schematic of major pathways associated with cell survival, differentiation, and proliferation.Arrows represent active signals and red lines represent inhibitory signals.mTOR: mammalian target of rapamycin; MITF:microphthalmia-associated transcription factor
PI3K-AKT pathway
The neuroblastoma RAS viral oncogene (NRAS) is mutated in approximately 30% of melanomas[168].Ras binds to PI3K activating secondary messenger PIP3 and binds to serine threonine kinase AKT[170].An important downstream effector of PI3K-AKT is mTOR [Figure 10], which functions to initiate a cascade that inhibits autophagy[160,171].PTEN is an antagonist of PI3K-mediated signaling.Loss of functional tumor suppressor genePTENincreases AKT phosphorylation leading to decreased apoptosis and increased melanoma survivability[165].PTENmutation was also found in nearly 44% ofBRAFmutated melanomas in contrast to 4% ofNRASmutated melanomas[168].These mutational patterns suggest thatBRAFandNRASmutations appear to be mutually exclusive and distinct oncogenic drivers[170].
c-KIT inhibition
c-KIT encodes for transmembrane tyrosine kinase KIT which binds to stem cell factor and activates cellular proliferation pathways including RAS-ERK and PI3K/AKT [Figure 10][162,172].KIT mutations were more frequently in mucosal, acral, and chronically sun-damaged skin melanoma than in non-chronic sundamaged skin melanoma in the United States[173].Of note, KIT mutations are believed to be more frequent in non-white populations and reported in higher mutation rates in East Asian patients[174,175].
Supporting stroma and vascularization
HIF-1α/VEGF
Tumors require vascularization to grow, metastasize, and invade.Angiogenesis correlates with the progression of melanoma tumor growth by supplying oxygen and nutrients and providing an opportunistic route to spread into blood circulation.VEGF is an important angiogenic factor and leads to secretion ofMMPs degrading extracellular matrix and allowing for openings for cells to invade neighboring tissues[174].HIF-1α initiates VEGF-A expression and key to downregulating mitochondrial energy metabolism by switching to glycolysis[163].This Warburg effect of lactic acidosis contributes to the acidification of the tumor microenvironment triggering metabolic reprogramming in melanoma cells [Figure 11] with increased proangiogenic VEGF and altered immunosuppression[167].

Figure 11.Multifactorial reprogramming to acidification of melanoma tumor microenvironment.The pH and oxygen microenvironment work together with genetic and metabolic pathways to promote melanoma proliferation and metastasis progression[176].HIF-1α:hypoxia-inducible factor-1 alpha; mTOR: mammalian target of rapamycin
Melanocyte-lineage
Microphthalmia-associated transcription factor
The microphthalmia-associated transcription factor (MITF) gene is a key regulator for the development and differentiation of melanocytes.Melanoma cell phenotype switching is characterized in the ERK/MAP-kinase pathway [Figure 10] targeting MITF phosphorylation in either transient or sustained activation[177].Sustained ERK phosphorylation inhibits MITF keeping its levels low leading to proliferation.High MITF levels along with transient ERK phosphorylation triggers melanocyte differentiation[178].cAMP signaling activates HIF-1α and increases MITF levels[179].HIF-1α and VEGF promotion triggers microenvironment changes that promote melanoma progression [Figure 11].Cellular crosstalk in signaling pathways also plays a driving role.MITF activates anti-apoptotic geneBCL2, promoting melanocyte survival[180].Cell cycle inhibitors p21 has been implicated along with p16INK4A and p14ARF [Figure 10] in the senescence of melanocytes[181].As the fate regulator of the melanocyte lineage, MITF activity fluctuates through modification of signaling pathways and microenvironmental dynamics.
Modulating immune response
Melanoma utilizes co-inhibitory pathways to avoid surveillance by the immune system and its ability to respond and possibly eradicate the tumor cells.Two well-characterized co-inhibitory pathways involve the cytotoxic T-lymphocyte associated antigen 4 (CTLA-4) and programmed death-1 (PD-1) T-cell co-receptor[182].CTLA-4 outcompetes CD28 for B7 binding on antigen presenting cells, limiting IL-2 production and lowered survivability of the T-cell[183].PD-1 is induced later during T-cell activation and ligation with PDL1 and PDL2 promote tumor growth[184].Immunotherapy for melanoma blocks PD-1 and CTLA-4 checkpoints and elicits a therapeutic response[183].The role of mechanical trauma in the development of melanoma remains unclear.One retrospective analysis on acral melanoma accounting for the majority of melanoma in the Chinese population provided epidemiological evidence for potential association between trauma and acral melanoma[185].For dark-skinned and East-Asian populations, patients have reported direct trauma related to subungual melanoma[186].However, there is not enough literature to support an established correlation.
Future directions
Many patients with good prognosis indicated by early stage through traditional clinicopathologic histology staging methods still decline from metastases[187].Recent advances in personalized medicine could be instrumental in capturing a more accurate and detailed micro-staging of melanoma through nextgeneration sequencing and genetic profiling.Validation studies of the 31-GEP, which categorizes risk as low or high by class, has indicated the 31-GEP test as an accurate predictor of metastasis of cutaneous melanoma[188-190].Systematic meta-analysis demonstrated the 31-GEP test consistently identifies melanoma patients at increased risk of metastasis independent of clinicopathologic factors and improves on current staging[191].The identification of early stage low-risk patientsvs.late stage high-risk stage patients would significantly help in determining life-saving adjuvant therapies or avoiding unnecessary treatments and costs.Analysis of profiles of gene expression modification induced by different signaling pathways in melanoma cells has considerable potential in understanding clinically relevant molecular mechanisms and biomarkers for non-melanoma skin cancers to create personalized treatment plans for each patient with melanoma.Unfortunately, the use of 31-GEP testing on thin melanomas is controversial.A recent article believes that the testing should not be done on people with thin melanomas if there is no evidence on change of outcome and treatment plan.Once further studies on 31-GEP testing are done, the evidence of effectiveness in thin melanomas will be apparent[192].
Summary
Recent endeavors into understanding the pathways involved in the development and metastasis of skin cancer into melanoma has led to some degree of hope in therapy against this historically difficult to treat cancer.It is clear melanoma is not homogenous, but a complex multifactorial disease characterized by crosstalk of several different pathways and elucidation of mechanisms that contribute to tumor growth and chemoresistance.Hence, understanding these complex mechanisms is crucial and will lead to more optimal treatments for each patient.
CUTANEOUS T-CELL LYMPHOMA
Cutaneous T-cell lymphoma (CTCL) is a type of T-cell mediated dermatologic neoplasm which falls under non-Hodgkin’s lymphoma[193].Mycosis fungoides (MF) and Sézary syndrome (SS), the most frequently diagnosed forms of CTCL, have become some of the most researched forms of non-melanoma skin cancers.CTCLs are often misdiagnosed as benign skin conditions, such as atopic dermatitis or psoriasis[194].Understanding the various signaling pathways of CTCL is conducive to forming proper treatment plans and prognoses.The goal of this section is to discuss the pathways in which CTCL arises and the treatment options utilized for this form of non-melanoma skin cancer.
There are various theorized and studied pathways in which CTCL arises.The majority of these cases do not progress to systemic malignancy, rather, it remains a mostly cutaneous cancer.Clinical features of CTCL can be fairly variable.From erythrodermic exanthems to flesh colored papules, CTCL can present in a number of cutaneous manifestations.There are a few exogenous factors to note that have been studied to have an antigen induced CTCL reaction.A common pathway in which CTCL evades the body is via the antigen-antibody mechanism.In the antigen-antibody mechanism, T-cells are consistently exposed to specific antigens resulting in proliferation and failure to induce cellular apoptosis[195].Not only are medications thought to be provoking factors in antigens causing T-cell proliferation, but viral, bacterial,and fungal antigens are also factors causing uncontrolled T-cell proliferation[195].Some of these viral antigens include human T-cell lymphoma virus, Epstein-Barr virus, and herpes simplex virus.In addition,methicillin resistantStaphylococcus aureus(MRSA),Mycobacterium leprae,Chlamydia pneumoniae,and even fungal dermatophytes can be inducing factors causing CTCL[196,197].Furthermore, there is an association between vitamin D deficiency, and even vitamin D receptor single nucleotide polymorphisms,causing deficiencies within the system.Vitamin D is important for the function of many immune cells including those of T cells.Paracrine communication between B and T lymphocytes,Cathledicingene regulation and innate immunity all function with the use of Vitamin D.Without properly regulated innate and adaptive immunity,Staphylococcus aureusand other bacteria and viruses have a higher probability of colonizing the skin while continuously priming and activating T lymphocytes.As mentioned earlier,Staphylococcus aureushas a known association with CTCL meaning that Vitamin D and polymorphisms in the VDR can indirectly have an association with CTCL[198].
Some studies have found association between atopy and CTCL with an IgE increase found in biopsies.However, there is a need for further research in the subject of atopy and allergen-induced CTCL.
Extrinsic exposures
Various studies have determined that if the antigen producing agent is removed, then the CTCL response will eventually eliminate itself.When comparing patients with CTCL and hypertension taking hydrochlorothiazide to those with CTCL that are not taking hydrochlorothiazide, complete clearance or improvement of their CTCL cutaneous lesions has been observed when hydrochlorothiazide is withdrawn[195].What is most important is that it is understood the different ways in which CTCL can be treated.Disruption of specific pathways via monoclonal antibody addition, as well as removing an antagonizing antigen, are just a couple examples in how this research has allowed for the elimination of this cutaneous neoplasm.
MRSA has also been found to be an insult leading to CTCL.Moreover, the treatment of MRSA can lead to improvement of CTCL.Patients with erythrodermic CTCL were discovered to have a decreased body surface area affected when being treated for a MRSA infection[196].This finding leads to the conclusion that patients with erythrodermic CTCL have obtained a coinfection MRSA.Once treated, the cutaneous manifestations dramatically decrease, thus, it is recommended that patients with CTCL be empirically treated for a MRSA infection[196].
While many of the signaling pathways discussed are being studied to better understand CTCL progression and initiation, there are various other pathways continuing to be discovered and evaluated in association with this cutaneous neoplastic disease.Cutaneous T-cell lymphoma is a result of malignant T-lymphocytes proliferation and invasion[194].
Apoptosis evasion
Epigenetics
Many of the identified common mutations in CTCL are in genes coding for proteins that alter epigenetic factors, including AT-Rich Interaction Domain 1A (ARID1A) andDNMT3A[199].ARID1Aalters histone structure, exerting its effects through epigenetic mechanisms.Mutations inARID1Ahave led to the downregulation ofPTENin other types of cancer[200].The effect ofARID1Adownregulation leads to decreased apoptosis and uncontrolled cell growth between the G1 and S phase of the cell cycle.DNMT3Ais a gene that encodes for DNA methyl transferase 3α, and mutations inDNMT3Aare linked to aberrant cytosine methylation and potentially the downregulation or silencing of important oncogenes[201].
One commonly researched target for CTCL is the NOTCH family of proteins.NOTCH1 activity is increased in both SS and tumor stage MF.The activity of NOTCH1 is typically upregulated through increased levels of binding ligand, Jagged1.Jagged1 ligand binds through a juxtacellular mechanism to NOTCH1.NOTCH1 is then cleaved into ICN1 by proteases.ICN1 exerts downstream effects, one of these being the expression of hairy and enhancer of split-1 (HES1) [Figure 12][202].In tumor stage MF, the suspected upregulated ligand is Jagged1.This is thought to occur through an epigenetic pathway involving DNA hypermethylation of the miR-200c promoter region[202].miR-200c can function as an oncogene that inhibits activation of NOTCH1 through decreasing expression of the NOTCH1 ligand Jagged1.However,hypermethylation in the promoter sequence of miR-200c decreases expression of miR-200c is seen in MF.This results in increased NOTCH1 activity through an increase in levels of Jagged1[202].Overactivity of NOTCH1 has been linked to decreased expression of PTEN and degradation of p53 in T-cell acute lymphoblastic leukemia[203].In addition, HES1 expression is also linked to the upregulation of the NF-κB pathway in T-cell acute lymphoblastic leukemia[204].There is a need for more research on the cause of hypermethylation of these miRNA regions that cause downregulation.
NOTCH1 is also linked to the AKT pathway through PTEN and p53 regulation by miR-122.miR-122 activates AKT[205].TP53 mutations are found in CTCL[199].miR-122 is primarily known for causing apoptosis within hepatocytes, yet recent studies have found that miR-122 is also found throughout human skin, especially in patients with MF.Lesioned skin has been found to express higher levels of miR-122 in patients with MF and SS compared to healthy skin.Though the normal function of miR-122 is found to induce apoptosis in hepatocytes, when upregulated, miR-122 blocks apoptosis.Cells with upregulation of miR-122 have been discovered to have phosphorylated AKT and downstream transcription factors in the pathway.As a result, AKT is then overactivated, leading to decreased apoptosis from increased TP53 inhibition.This lack of apoptosis leads to the uncontrolled cell growth of cutaneous T-cells[205].
Malignant cell growth
Mutations upregulating the Janus kinase (JAK) and signal transducer and activator of transcription proteins (STAT) are implicated in CTCL.JAK1, JAK3, STAT3, and STAT5B are mutated in certain CTCL cell lines[206].Mutations in JAK1, JAK3, and STAT5B have also been implicated in T-cell prolymphocytic leukemia[207].The effects of JAK/STAT signaling in CTCL are thought to be driven partially by the production of cytokines and angiogenic factors[194].Generally, the mechanism of JAK/STAT signaling is as follows.Specific ligands or cytokines bind to the RTK, causing activation and dimerization of the receptor.This dimerization allows JAK proteins to come into close proximity to each other.JAK-JAK phosphorylation occurs, and JAK then phosphorylates STAT proteins.STAT proteins then dimerize and translocate to the nucleus, where they act as transcription factors [Figure 12][208].

Figure 12.Common pathways in cutaneous T-cell lymphoma.This figure describes the genetic signaling pathway of NOTCH1.NF-κB:nuclear factor-kappa B; JAK: Janus kinase
Activated JAK is also known to activate PI3K, resulting in eventual activation of AKT [Figure 12][208].Activation of AKT has also been implicated to occur in CTCL through mutations in the PI3K pathway relating to TCR-CD28[209].Additionally, loss of heterozygosity in PTEN, the negative regulator of PI3K, have been observed in MF, and PTEN activity is shown to be altered in CTCL[194].The PI3K pathway is explained in greater detail in the SCC section of this paper.Activated JAK is also known to activate PI3K, resulting in eventual activation of AKT [Figure 12][208].Activation of AKT has also been implicated to occur in CTCL through mutations in the PI3K pathway relating to TCR-CD28[209].
NF-κB pathway activity has also been shown to be increased in CTCL[206].The canonical NF-κB pathway occurs as follows.IkB kinase (IKK) acts to phosphorylate the IkB element of NF-κB-IKB.Phosphorylated IkB is ubiquitinated, which allows for the degradation of IkB by proteasomes.NF-κB is then able to enter the nucleus to act as a transcription factor[204].One of the effects of NF-κB signaling is an upregulation of its negative regulator IkB.This serves as a negative feedback mechanism of NF-κB signaling [Figure 12][210].The NF-κB pathway is also linked to the activation of AKT.One of the actions of AKT is the phosphorylation of IKKα, which is part of the IKK complex of the canonical NF-κB pathway [Figure 12].
The non-canonical NF-κB pathway is implicated also in CTCL.Mutations are seen in the C terminal of the geneNFκB2, which is an area encoding for autoinhibitory functions in NF-κB2.Typically, the C terminal of inactive NF-κB2 assists in suppressing the canonical NF-κB pathway.Mutations in the NFκB2 gene are,therefore, thought to contribute to activation of the canonical NF-κB pathway in CTCL[199].
The genesphospholipase C, gamma 1(PLGC1), andcaspase recruitment domain family member 11(CARD11) are amplified in CTCL and are thought to contribute to abnormal TCR signaling[199].PLGC1activates the NF-κB pathway and nuclear factor of activated T-cells through diacylglycerol and inositol 1,4,5-trisphosphate (IP3), respectively[211].CARD11is linked to the induction of the NF-κB pathway,although the mechanism of action is not yet completely understood[212].
Other mutations in tumor suppressor genes,CDKN2A,TNF-α Induced Protein 3(TNFAIP3),zinc finger E-box binding homeobox 1(ZEB1) andFAS, have been identified in CTCL[199].TNFAIP3 protein works to inactivate IKK when the NF-κB pathway is overactivated by TCR[213].ZEB1 is a zinc finger transcription repressor protein, and the loss of ZEB1 in CTCL is linked to the overexpression of cytokines like IL-2.Mutations inZEB1also increases the expression ofGATA3, which binds to the promoter site of the gene encoding for thymocyte selection-associated HMG box protein (TOX)[199,214].TOX is a mediator of T-cell maturation from CD4+and CD8+to CD4+[214].The loss of FAS in CTCL cell lines most likely also contributes to apoptosis resistance[199].
As mentioned previously, the pathogenesis and progression of CTCL are believed to be influenced by alterations to the local cytokine milieu.These changes are thought to occur in part due to disfunction in CXCR4[215].CXCR4 mediates downstream effects of various receptor tyrosine kinases, one of which is TCR.When TCR is activated, CXCR4 is able to ligate with TCR, triggering a process that enhances the stability of mRNA transcripts for IL-2, IL-4, and IL-10.This process occurs in the presence and in the absence of CXCL12, the sole endogenous ligand for CXCR4.Furthermore, blocking the association between CXCR4 and TCR decreases mRNA transcript stability for both TCR-dependent and TCR-independent cytokine production in SS cell lines, without altering other downstream effects of TCR[215].
Supporting stroma and vascularization
More recently, CTCL progression via angiogenesis is being studied.Angiogenesis via the VEGF pathway results in the congregation of tumor cells[216].The stromal involvement of CTCL involves a CXCL12[216].The stromal factor and its adjacent receptor work to create a signaling network for bone marrow stem cells,which is found in earlier disease states[216].
Summary
Though there is a need for further research on CTCL, there are many treatment options available.Historically, first line treatment for CTCL has been psoralen and UVA (PUVA) therapy[217].When used in conjunction with interferon-α, efficacy has been shown to increase due to elevated levels of apoptosis[217].PUVA upregulates p53 allowing increased apoptosis within the tumor cells[217].With the addition of interferon-α, apoptosis is increasingly regulated via the JAK1 signaling pathway[217].
Brentuximab vedotin is also a potential treatment for CTCL[218].This monoclonal antibody has been shown to have efficacy against refractory cases of CTCL, as well as peripheral T-cell lymphoma[218].During the studies performed using this medication, it was found that success is greatest in those CTCL cases where CD30+positive cells are present.CD30+falls within the TNF receptor class, which is involved in cell proliferation[218].Thus, explaining the mechanism of this monoclonal antibody to inhibit specifically CD30+expression[218].Another monoclonal antibody used in the treatment of MF and SS is Alemtuzumab[219].Thus, research of immunomodulatory treatments is on the rise for CTCL leading to personalized treatments targeted to the genetic signaling pathways, which are dependent on the person’s specific flux state.
CONCLUSION
Melanoma and non-melanoma skin cancer are some of the most prevalent cancers that can lead to death if not properly managed.However, knowing the genetic signaling pathways of these conditions can further help researchers and providers give more options to patients suffering from these conditions.With this information, the provoking genetic factors can be blocked with the use of pharmaco- and immunotherapy allowing for patients with treatment resistant, locally advanced and even metastatic lesions to be treated due to the different options of therapy blocking multiple signaling pathways.Though some information is known about the genetic pathways of cutaneous malignancies, there is still a demand for research that can be done to improve treatment options to decrease mortality rate and recurrence.
Initiators and promoters
There are different provoking aspects that cause cutaneous malignancies.Promoters are products or a lowlevel exposure a carcinogen that can begin the process of tumorigenesis but not quite reach the threshold to create full DNA mutations.Exposome is the combined exposure to all factors that cause internal chemical environment change[220].Promoters, however, can change gene expression that leads uncontrolled growth.Initiators are factors that begin the process of DNA mutation and alteration in structure.It is not the exposure to one carcinogenic chemical or product that immediately causes DNA mutation and tumorigenesis, but it is the initial exposure that primes the tumor cells to change the function of certain genes.Eventually, the exposome can promote tumors to form[220].It is also not just the exposure to one carcinogen in low doses but the repeated exposure to multiple carcinogens.In the case of cutaneous malignancies, solar UV is seen as a carcinogen that can either work as a promoter, an initiator, or both a promoter and initiator depending on the amount of exposure and protective measures.Moreover, studies have found that children exposed to the high-level UV from Western Australian sun have been found to have varying risk of cutaneous malignancies.Patients that arrive to Western Australia prior to age 10 have a 50% increased risk of dysplastic nevi or melanoma compared to those who arrive after the age of 10 regardless of their time spent sunbathing.This factor is due to the solar UV in Western Australia being higher of many other countries from where patients immigrate.Solar UV acting as an initiator and/or promoter for cutaneous malignancies explain the importance of protecting the skin from solar UV at all costs regardless of Fitzpatrick skin type.There is a need for further research to find other carcinogens that can act as initiators and/or promoters that to help patients prevent from these cancer-causing factors[221].
Effects of mi-RNAs
Genetic testing and gene therapy might be the future for patients with cutaneous malignancies.As previously stated, Castle Biosciences has already created testing to further determine staging and implication for sentinel node testing for melanoma and metastatic risk for SCC.However, the genetic testing does not stop there.miRNA plays an important role in signaling pathways determining the activation and suppression of specific genes for transcription factors.As mentioned in the previous sections, miRNAs can cause hypomethylation of genes leading to increased transcription, translation, and uncontrolled proliferation.Many of these genes are upregulated and downregulated having different effects on the genes; these effects can also lead to the appearance and progression of these cutaneous cancers.Moreover, SCC, VC, and CTCL have all been found to be incited by miRNAs.There are hundreds to thousands of miRNAs that can transform the function of genetic pathways and transcription factors from a normal functioning pathway to a malignant pathway.With this information, miRNAs can be a subject of research to help further test patients and possibly place them in complete remission as well as prophylaxis to prevent the next cutaneous malignancy from appearing.
Further genetic testing
In addition to genetic testing and therapy, there are many more topics within genetics that have room for further research.Many of the genetic tests performed are through somatic genetic testing and germline genetic testing.The germline testing is done through either saliva or lymphocytes.However, studies have found that within the saliva of patients with systematic lupus erythematosus, their salivary DNA has adapted the DNA of their food products[222].“You are what you eat”.This information can be helpful;however, with determining the somatic genetic susceptibility of cutaneous malignancies, there must be a way to determine the difference between the genetic makeup of the food consumed and that of the patient’s unmutated, unincorporated somatic cell DNA and RNA.This can be a problem with lymphocytes as well.T- and B-lymphocytes have specific genetic makeup that turns them into the subtype of mature T- and B-lymphocytes.Yet, when T- and B-lymphocytes are presented with a specific antigen or allergen, their genetic makeup can also transform to program the lymphocyte for either attack of the antigen or produce antibodies towards the antigen, respectively.Additionally, lymphocytes can change their genetic codedepending on their antigen priming, thus cancer cells may promote a change in lymphocyte DNA to avoid recognizing the cells as foreign[223].There also will need to be consideration for the use of inactive mature B- and T-lymphocytes that have not yet been presented with antigen compared to that of the lymphocytes that have been presented with an antigen.It is possible that the human body is a state of active dynamic mosaicism with genes turning on and off that have adapted genetic material to food products and other antigens within the body.This information can ensure further accuracy when determining the somatic genetics of patients with cutaneous malignancies and help with their treatment and prognosis.
Cellular electrical charges
Another important factor to consider with skin cancer genetics is that tumors tend to also have an electrical charge as do many cells within the body.This is also a way to evade immune system attack.Tumor cells are negatively charged.Lymphocytes that have been primed with tumor antigen are also negatively charge.In the world of chemistry, similar charges are opposed to each other while opposite charges attract each other.However, antibodies are positively charged.The positive charge of antibodies assists with T-helper lymphocyte eradication of tumor cells.However, it is known that natural killer cells and CTLs are important in immune killing of tumor cells, so there is more research that needs to be done in terms of the effect of charge between immune cells and tumor cells as well as on genetic signaling.
Communication between immune system and nervous system
The central and peripheral nervous systems are important for regulating the systems in the body.In fact,the nervous system directly affects the function of the immune system against fighting malignancies.Chronic exposure to stressful events stimulates the adrenal gland, a neuroendocrine organ, to release sympathetic nervous system stimulating hormones which disturbs the normal function and balance of the immune system.In addition, this stress can also increase lymphatic vascularization in current tumors that increases the number of chemokines release and promotes diapedesis of tumor cells.It is important to note and follow patients who have chronic stressors without stress relief exercises to maintain homeostasis.This neuroendocrine modulation also can influence the appearance of cutaneous tumors.As a result, decreasing stressful events in the lives of patients can help prevent cutaneous malignancies from occurring[224].
The body as a flux state
It is important to remember that tumors are not always a collection of cells that function the same way undergoing the same processes at the same time.Many of these tumors function as organs, like that of the heart or lungs with various cell types and cell functions.Likewise, there can be tumor cells that are evading apoptosis by downregulating tumor suppressors, other cancer cells that are promoting cell growth through the mutation of oncogenes while others are invading the stroma by damaging the underlying adhesive molecules and releasing proteinases.In addition, cancer cells also can communicate amongst each other with autocrine and paracrine signaling.This signaling can activate specific genetic pathways to even mutate a normal, non-cancerous cell to that of a cancerous cell and change the genetic pathway of those that are already malignant.All these processes can occur simultaneously especially in the faster growing, more aggressive types of skin cancer.This hypothesis can explain why there are patients that can go into complete remission from a treatment type and others are treatment resistant.The multiple genetic signaling pathways activated, crosstalk and switching from one genetic pathway to another can be the reason as to why there is treatment resistance.Tumor cells, like immune cells, may have an ability to release chemokines leading to autocrine and paracrine communication between the cells.This process can also form a communication system between the cells telling each of them their function and even invade benign cells to become malignant.Adjuvant therapy might be considered in the earlier stages of cutaneous malignancies depending on the number of genetic pathways that are discovered within these tumor cells.These discoveries give providers further information to continuously perform full body skin examinations and surveillance patient chronic inflammatory conditions for transformation into malignant conditions.Furthermore, the incorporation of artificial intelligence data analysis of trends between the various pathways can help improve management of cutaneous malignancies by clinicians.Once these conditions are elucidated, oncologists can develop prospective personalized dynamic genetic testing for identifying the critical set of active oncogenetic pathways that need to be disrupted at particular moments.This process would then transform the lives of individual patients suffering from multiple cutaneous malignancies around the world.
DECLARATIONS
Authors’ contributions
Wrote, final edited and revised the manuscript: Maner BS, Dupuis L, Su A, Jueng JJ, Harding TP,Meisenheimer VII J, Siddiqui FS, Hardack MR, Aneja S, Solomon JA
Contributed to Figure 10: Su A
Contributed to Figures 1, 2, 3, 6, 7, 8, 12 and reference organization: Meisenheimer VII J
Availability of data and materials
Not applicable.
Financial support and sponsorship
None.
Conflicts of interest
None that may be perceived as influencing the representation or interpretation of review other than Dr.James A.Solomon was a clinical investigator of Vismodegib clinical trials and expanded access programfunds paid to his employer and has been a paid consultant by Genentech, Sun Pharmaceuticals, Mayne Pharmaceuticals.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2020.
 Journal of Cancer Metastasis and Treatment2020年9期
Journal of Cancer Metastasis and Treatment2020年9期
- Journal of Cancer Metastasis and Treatment的其它文章
- Liquid biopsy in endometrial cancer
- Epithelial-mesenchymal transition: a hallmark in pancreatic cancer stem cell migration, metastasis formation, and drug resistance
- The good and bad sides of exosomes: premetastatic niche formation, cancer biomarker and therapy carriers
- Role of autophagic response induced by major phytochemicals in cancer prevention and treatment
- Extracellular RNAs as potential biomarkers for cancer
- Exploiting the relevance of CA 19-9 in pancreatic cancer