
Role of autophagic response induced by major phytochemicals in cancer prevention and treatment
2020-07-20 03:48:10ColeWennerRyanStewartAbigailMcCabeJasonPepeJohnOberlinAdamVanWertAjayBommareddy
Cole Wenner, Ryan Stewart, Abigail McCabe, Jason Pepe, John Oberlin, Adam VanWert, Ajay Bommareddy
Department of Pharmaceutical Sciences, Nesbitt School of Pharmacy, Wilkes University, Wilkes-Barre, PA 18766, USA.
#Equal author contribution.
Abstract Phytochemicals derived from dietary sources and natural products have gained significant attention in the scientific community due to their ability to modulate various pharmacological and biological activities.Understanding the molecular mechanisms by which natural products protect against various diseases including cancer will provide the basis for both clinical use and further chemical modification to develop targeted therapy.Autophagy, an evolutionarily conserved self-digestion process that employs lysosomal-mediated enzymatic degradation has a functional role in a wide range of pathological disorders, and has attracted oncology scientists over the past two decades.Studies employing natural products have shown that induction of autophagy may be either cytoprotective or cytotoxic governed by different molecular pathways.In this review, we summarize four major phytochemicals namely phenethyl isothiocyanate, capsaicin, withaferin A, genistein and their association with autophagy in cancer chemoprevention.We also discuss ideas for further investigation essential to understanding their mechanisms, which will guide their clinical applications for cancer prevention and treatment.
Keywords: Cancer, autophagy, capsaicin, phenethyl isothiocyanate, genistein, withaferin A
INTRODUCTION
Autophagy/macroautophagy is an evolutionarily conserved catabolic process that provides energy and macromolecular precursors through lysosomal degradation of cellular material[1,2].Autophagy occurs at an elementary level in all eukaryotic cells during their normal growth and development.Although the role of autophagy in energy homeostasis is quintessential, its role in cellular processes such as degradation of cytoplasmic organelles and proteins is also well established.The process of autophagy typically begins with the formation of a phagophore, followed by recruitment and processing of light chain 3 I (LC3) to LC3-II occurring in the presence of an autophagic stimulus with eventual sequestration into an autophagosome.Subsequently, the newly developed autophagosome engulfs cellular proteins and organelles, fuses with a lysosome, forming an autolysosome, where autophagy occurs[2].Intracellular components including mitochondria and endoplasmic reticulum are engulfed by the newly formed phagophore before undergoing degradation upon fusion with lysosomes.This process facilitates the recycling of bioactive ingredients for cellular sustenance[3-5].The degradative products, which include simple carbohydrates and amino acids are recycled back into the cytoplasm.The mammalian target of rapamycin (mTOR), a highly conserved serine/threonine kinase senses signals from growth factors, energy status and stressful environment and serves as a key regulator of cell growth and division.mTOR’s ability in mediating cellular responses is known to depend on the availability of amino acids and growth factors.It is usually active in an amino acid-rich environment and usually regulates protein translation but inhibits autophagy.When the levels of extracellular amino acids are very low, the process of autophagy recycles intracellular components to restore amino acid levels[6].Upregulation of mTOR results in excessive cell proliferation and eventually leads to the development of cancerous growth.Inhibition of mTOR results in induction of autophagy.The deregulation of autophagy has been clearly established in various chronic conditions including cancer.On the basis of the stimuli and tumor type, autophagy has been reported to be tumor promoting, or tumorsuppressive, indicative of the context-dependent role of autophagy in cancer[4,5].Studies also suggest that pharmacological or genetic inhibition of the autophagic response increases the chemotherapeutic efficacy of conventional agents, suggesting that autophagy inhibition in situations where it promotes cell survival may be an appropriate strategy in the treatment of cancer.
A large body of evidence including several epidemiological studies shows an inverse association of a diet rich in fruits and vegetables and the development of various cancers[7-9].In the last couple of decades,scientists and especiallycancer biologists have started exploring the role of major dietary phytochemicals consumed by people across the world.Studies have identified the ability of these phytochemicals to modulate important signaling pathways and oncogenes associated with cancer development and progression.To that end, various studies have also looked into the role of plant-derived phytochemicals in the autophagic response and have reported important findings in their ability to modulate this cellular process as a way to affect the development of chronic conditions including cancer[10-12].However, further research is essential to understand the mechanistic details associated with induction and/or inhibition of autophagy by these bioactive compounds.The current review summarizes established findings related to the autophagic response to four major phytochemicalswidely consumed by different cultures across the world.
PHENETHYL ISOTHIOCYANATE
Phenethyl isothiocyanate (PEITC) is a naturally occurring isothiocyanate containing a phenethyl group attached to its nitrogen.PEITC is found in various cruciferous vegetables in the form of gluconasturtiin.Myrosinase, an important enzyme located in the cellular matrix, catalyzes the conversion of PEITC from glucoasturtiin.When cruciferous vegetables, such as watercress, broccoli, radishes, or turnips are chewed or crushed, myrosinase is activated[13].Myrosinase can also be activated in the digestive system,which allows PEITC to be released after the vegetable has been ingested.Both myrosinase and PEITCare thermolabile, and therefore cooked forms of cruciferous vegetables provide considerably less PEITC to the body than raw forms[13].Recent studies of PEITC have focused on the phytochemical’s possible role in cancer chemoprevention.Specifically, prostate cancer prevention has received attention, owing to its long latency and because dietary exposure to PEITC is noninvasive and rather promising.Androgen receptor signaling plays a significant role in many aspects of prostate cancer cell growth[14], suggesting the utility of androgen responsive LNCaP cells, which are commonly used in many different prostate cancer studies[15].The mitochondria’s role in metabolism makes it a major source of cellular energy.Therefore,inhibiting mitochondrial function also depletes the available cellular energy, forcing the cells to destroy themselves and use their contents for energy[16].Studies explored the mitochondrial structural alterations and formation of autophagosomes upon treatment with PEITC in prostate cancer cells[17,18].PEITC-induced autophagyalong with apoptosis, contributing to growth suppression of prostate cancer cells both in cell culture and in a transgenic mouse model of prostate cancer[19,20].
Glycolysis refers to the aerobic or anaerobic catabolism of glucose, which results in pyruvate, NADH,and ATP.Certain metabolic intermediates of glycolysis can also benefit cancer cells, as they promote macromolecular biosynthesis, which enhances the available nutrient supply[21].Without the products of glycolysis, cancer cells utilize the autophagic response, where they degrade cytosolic organelles to generate energyfor survival[22].The effect of PEITC on glycolysis, miRNA, and the matrix metalloprotease (MMP),MMP2/MMP9 signaling pathway was shown to slow the spread of prostate cancer.This study further explored how PEITC could affect c-Myc-regulated glycolysis.PEITC exposure increased the level of c-Myc proteins, known to inhibit the expression of the enzymeshexokinase 2 (HKII), pyruvate kinase isozyme 2(PKM2), and lactate dehydrogenase A (LDHA), whichpromote glycolysis[23].PEITC exposure was shown to inhibit glycolysis, as revealed by suppressed expression of important markers including HKII, PKM2,and LDHA in LNCaP and 22Rv1 cells[23].This is a revolutionary finding, as PEITC-mediated inhibition of glycolysis in cancer cells would eliminate a major energy store for prostate cancer cells, allowing for a new type of chemotherapy, utilizing the cell’s natural autophagic response system.In addition, by inhibiting glycolysis in cancer cells, PEITC has the potential to eliminate the common problem of multidrug-resistant(MDR) hypoxia[24,25], which is a major complication for other chemopreventive treatments.Hypoxia, a known factor that transforms cancer cells into MDR cancer cells, also promotes cellular glycolysis through what is known as the Pasteur effect.Hypoxia does this by manipulating cancer cells’ metabolism, resulting in the induction of cellular quiescence[25].Therefore, hypoxia is a major cause of chemotherapeutic drug resistance.Thus, PEITC’s role in inhibiting glycolysis in cancer cells may limit the hypoxic environment and minimize the transformation of MDR cancer cells.
PEITC was also shown to induce autophagic response through the regulation of MMP2/MMP9 signaling pathways.Both MMP2 and MMP9 proteins are responsible for extracellular matrix collagen/protein degradation.Activation of MMP proteins can promote angiogenesis and limit cellular apoptosis, allowing cancers to proliferate[26,27].MMP2, MMP9, and c-myc work together in cancer progression.PEITC was shown to downregulate the expression of MMP2, MMP9and c-myc proteins and simultaneously induceautophagy[26].A study using hepatoma-derived growth factor resulted in tumor growth inhibition,and proved a linkage between autophagy and MMP2, MMP9, and c-myc proteins.It also proved that by modulating the signal pathway of the MMP2 and MMP9 proteins, prostate cancer cell migration and invasion could be inhibited[28].MMP2 is also a downstream effector of the JAK/STAT pathway, involved in tumorigenesis and metastasis.PEITC was shown to suppress the JAK/STAT-MMP2 pathway and induce autophagyin vitroand in a lung cancer mouse xenograftmodel.However, autophagy induced by PEITC preserved metastasis potential, which was abrogated in the presence of chloroquine[29].A different study that explored PEITC’s epigenetic regulatory effects on miRNA in prostate cancer cells found that PEITC influenced miR-194 levels and downregulated the expression of oncogenic MMP2 and MMP9 proteins.The downregulation of MMP2 and MMP9 proteins ultimately reduced prostate cancer metastasis[30].It hasalso been shown that MMP2 and MMP9 proteins are both highly expressed in breast cancer tissues and are related to lymph node metastasis and tumor staging[27].These results further prove that the regulation of miR-194 and MMP2 and MMP9 proteins is critical in cancer research and that PEITC’sability to modulate the expression of these factors is vital in the induction of autophagy and prevention of metastasis.Table 1 summarizes the PEITC-induced autophagic response in different cancer models.
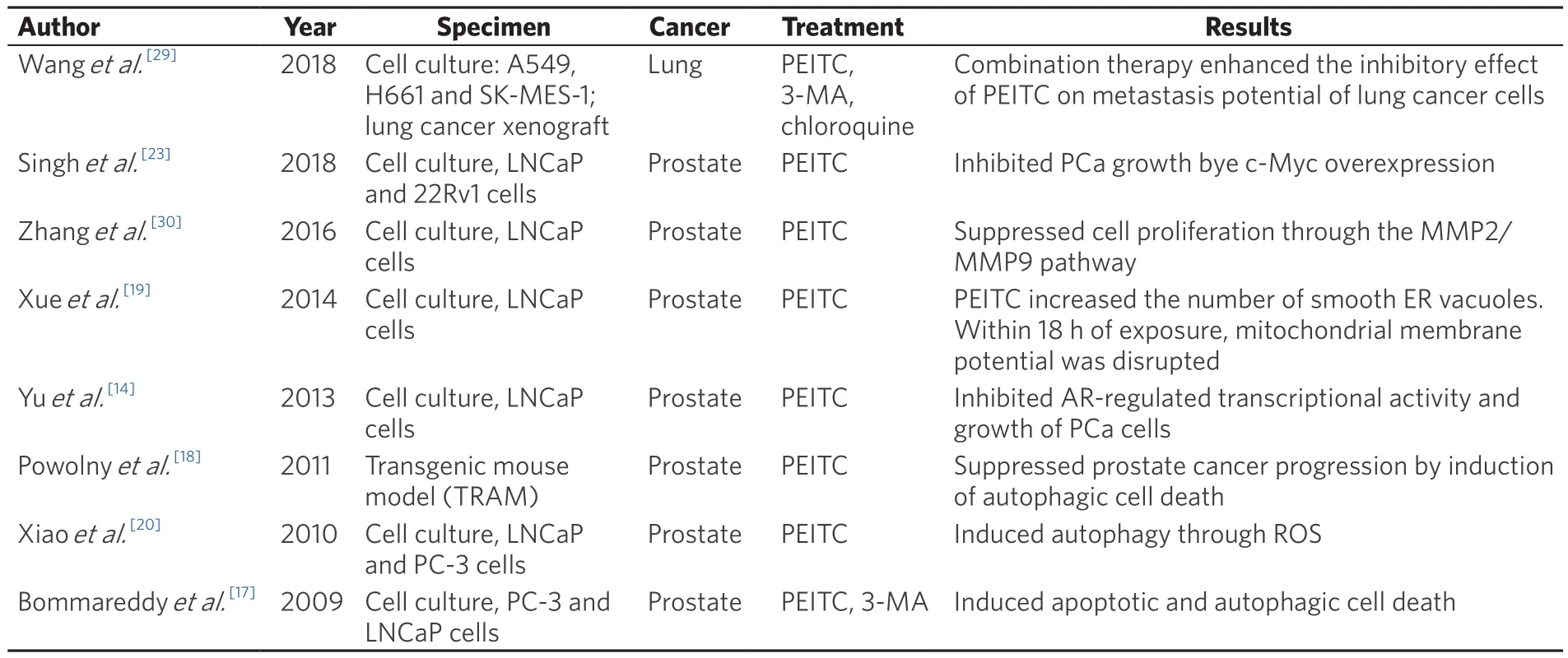
Table 1.Summary of autophagic response induced by PEITC in various cancer models
CAPSAICIN
Plants belonging to the genusCapsicumsynthesize the alkaloid capsaicin (CAP), known to give chili peppers their hot, spicy flavor[31].Despite being an important cultural ingredient in many cuisines,CAP possesses anticancer properties, which have become an area of scientific interest in regard to chemoprevention.Studies have aimed at elucidating the mechanisms in which CAP works to promote autophagy in cancer cells to foster clinical applications for cancer treatment.
Researchers have discovered that coupling CAP with other chemotherapeutic drugs can minimize cellular chemoresistance to conventional therapies.A study investigating the combination of CAP and cisplatin against human osteosarcoma (HOS) cells showed that CAP at lower concentrations and cisplatin displayed a synergistic reduction of cellular viability in MG63, 143B and HOS cell lines[32].This study further showed cell cycle arrest in the G0/G1 phaseand also examined the association of MMPs with inhibition of cellular invasion.The combination treatment resulted in decreased expression of MMP-2 and MMP-9 and reduced gelatin degradation by these enzymes.The study also investigated whether CAP/cisplatin treatment induced autophagy.Expression of the autophagy-associated proteins Beclin 1, Atg3, Atg5, Atg16,and LC3-II was increased along with the accumulation of autophagic vacuoles in the cytoplasm, indicating autophagy induction.CAP/cisplatin treatment was shown to target the reactive oxygen species (ROS)/AKT/mTOR pathway, a crucial mediator of cellular autophagy, by reducing p-ATK and p-mTOR levels.Upon treatment with the autophagy inhibitor bafilomycin (BAF), cells exhibited upregulation of apoptotic genes, suggesting that autophagy may be a pro-survival mechanism in osteosarcoma (OS) cells.CAP/cisplatin treatment significantly increasedthe generation of ROS, found to mediate the activation of the JNK pathway.In addition to reducing the cytotoxic effects of the combination treatment, pretreatment of HOS cells with N-acetyl cysteine (NAC), an ROS scavenger, reduced JNK phosphorylation and partially suppressed autophagy by reversing p-ATK and p-mTOR reduction[32].Another study examined the effects of CAP on the sensitivity of cholangiocarcinoma (CCA) cells to common chemotherapeutic drugs.TheCCA cellsexamined, QBC939, SK-ChA-1, and MZ-ChA-1, exhibited MDR to the chemotherapeutic drugs cisplatin, vincristine, and 5-fluorouracil (5-FU) as well as CAP.In vitro, combined treatment of 5-FU with low concentrations of CAP displayed synergistic effects in 5-FU-induced anti-proliferation, increasing sensitivity to apoptosis.The autophagy inhibitor 3-methyladenine (3-MA) enhanced the effectiveness of 5-FU inQBC939 cells, while the autophagy activator retinoic acid receptor alpha (RARA) promoted resistance to 5-FU.Studies have shown that the growth and survival effects of autophagy typically promote chemotherapeutic drug resistance.5-FU treatment increased the expression of Beclin 1, LC3-II, and Atg5 genes as evidenced by acridine orange staining, PCR, and Western blot analysis.CAP/5-FU co-treatment inhibited 5-FU-mediated autophagy through phosphorylation of the Akt/mTOR pathway.Hence, CAP hindered chemotherapeutic drug resistance, proving its potential as an adjunct to conventional therapies[33].
In response to environmental stresses such as DNA damage, CAP increases cellular viability through autophagy in certain cell lines.One study explored the impact of CAP on glioblastoma and breast cancer cell lines with mutated p53 oncosuppressor genes.Mutated p53 cells are known to express resistance to common therapies and promote tumor growth.The study noted that CAP increased levels of LC3-II,reduced p62 protein, and reduced mutant p53 by autophagy-mediated protein degradation.In addition,treatment caused p53 to transactivate damage-regulate autophagy modulator, a gene that induces autophagy through lysosomal protein expression, among other apoptotic genes.Western blotting confirmed that poly ADP-ribose polymerase (PARP) cleavage occurred after CAP treatment in both U373 and DKBR3 cells.The loss of mutant p53 inevitably altered the ratio of folded to misfolded p53 proteins, consequently restoring wild-type p53 protein activity in target cells[34].CAP-induced autophagy has also been shown to repair DNA in MCF-7 breast cancer cells.Autophagy was initiated by the AMPKα-mTOR pathway and the concentration of p53 protein increased in both the nucleus and cytosol.Moreover, cells experienced an alteration of mitochondrial membrane potential and blockingS-phase.In response to cancerous gene damage, CAP initiated DNA repair by activating δ-H2AX, ATM, DNA-PKcs, and PARP-1.Inactivation of the Atg5 gene in M059K cells resulted in phosphorylation of ATM and DNA-PKcs and increased expression of p53 and LC3-II.ATM and p53 inhibition resulted in PARP cleavage and prevented DNA-PKcs phosphorylation without changing LC3-II levels.Therefore, autophagy inducedby genetic damage improved cellular viability by repairing DNA through ATM regulation of DNA-PKcs and PARP-1[35].
Inhibition of CAP-induced autophagy can enhance cellular sensitivity to apoptosis in a variety of human cancers.CAP decreased cellular viability in U251 glioma cells by increasing the expression of Beclin1, p62, and PUMA.Treatment with 3-MA, a known inhibitor of PI3K that blocks the formation of autophagosomes, reduced procaspase-3 and Beclin1 while increasing the expression of p53 and PUMA.Inhibition of p53 increased the expression of autophagic proteins.Therefore, inhibition of CAP-induced autophagy in glioma cells contributed to apoptosis[36].As discussed above, CAP synergistically restricted proliferation, increasing CCA cell sensitivity to apoptosis and upregulated apoptotic gene expression upon autophagy inhibition in OS cells[32,33].Multiple studies have demonstrated that CAP promotes apoptosis in a time-dependent manner upon inhibition of autophagy in hepatocellular carcinoma, melanoma and bladder cancercells[37-39].CAP was shown to target the ROS/signal transducer and activator of transcription 3 (Stat3)pathway in hepatocellular carcinoma cells.Western blotting indicated increased expression of autophagy markers LC3-II and Beclin1, GFP-LC3 autophagosome formation, ROS generation, and upregulated phosphorylation of Stat3, a known regulator of autophagy.In addition, NAC reversed the effects of CAP on ROS/Stat3-dependent autophagy, consequently inducing apoptosis[36].CAP stimulated LC3-II levels,formation of acidic vesicular organelles, and expression of ubiquitin-binding protein p62, which transports other proteins to the phagophore complex for selective autophagy, in 5,637 and T24 bladder cancer cells.To test if CAP could induce autophagy at varying levels of p62, cells were co-treated with CAP and BAF,a lysosomal inhibitor.Cells were found to generate p62, proving that CAP can activate autophagic flux.It was noted that CAP treatment generated ROS production resulting in time-dependent depolarization ofthe mitochondrial membrane potential and an increase in the ADP/ATP ratio, both indicative of decreased cellular metabolic energy and autophagy.In addition, CAP increased concentrations of Atg4C, a protein responsible for autophagosome development and upregulated autophagic genes: GABA Type A Receptor Associated Protein Like 1 (GABARAPL1), LC3B, Sequestosome-1 (SQSTM1), immunity related GTPase M(IRGM), Unc-51 like autophagy activating kinase 1 (ULK1), tumor necrosis factor (TNF) and phosphatase and tensin homolog (PTEN).CAP-resistant cells underwent epithelial mesenchymal transition (EMT) and autophagy through activation of the Hedgehog pathway when exposed to CAP.Interestingly, activation of this pathway caused CAP-induced EMT cells to develop chemotherapeutic drug resistance to mitomycin C,gemcitabine, and doxorubicin[39].
Studies have indicated that multiple signaling pathways mediate CAP-induced autophagy.Dihydrocapsaicin(DHC), an analog of capsaicin, mediated autophagy in a catalase-dependent manner.Treatment upregulated the expression of Atg5, a gene necessary for autophagosome formation, in addition to Atg4 and Atg7 consequently increasing LC3-I conversion to LC3-II.Catalase induction by DHC decreased baseline levels of ROS, consequently increasing production of LC3-II protein and caspase-3 activation.Upon exposure to 3-amino-1, 2, 4-triazole (3AT), a catalase inhibitor, LC3-II levels from DHC treatment were decreased.Conversely, overexpression of the catalase gene resulted in increased expression of LC3-II[40].Lysosomes co-localized with LC3-II to form autolysosomes in treated LNCaP and PC-3 cells; however,blockade of autophagy prevented their formation.High intracellular levels of ROS were observed in LNCaP cells compared to PC-3 cells.Upon treatment with NAC, ROS production by CAP was decreased in both cell lines.Confocal microscopy confirmed that cells treated with NAC did not accumulate autolysosomes and reduced inhibition of the PI3K/Akt pathway, indicating the critical role of ROS in autophagy[41].Another study confirmed that treated NPC-TW01 cells displayed reduced interaction between Beclin1 and Bcl-2, suggesting that autophagy activation may be regulated by the Beclin1/Bcl-2 complex and the class III PI3K/Beclin1/Bcl-2 pathway.Inhibition of the PI3K/Akt/mTOR pathway suggested a correlation between CAP and cellular proliferation[42].Table 2 summarizes the capsaicin-induced autophagic response in different cancer models.
WITHAFERIN A
Withaferin A (WA), a steroidal lactone, is derived from a number of plants belonging to the family Solanaceae, includingAcnistus arborescensandWithania somnifera.It has been used in Indian medicine for centuries and is currently being investigated in the Western world for its anticancer, antitumor, and antiinflammatory properties; however, its mechanism of action is unclear and is currently being investigated.
ROS generation was shown to be one of the main mechanisms by which WA prevents the growth of cancer cells.For example, WA used alone or in combination with cisplatin has proven to induce cell death by generating ROS and subsequent DNA damage in ovarian cancer and non-small cell lung cancer (NSCLC)cells[43,44].The combination of doxorubicinand WA in the treatment of ovarian cancer cell lines A2780,A2780/CP70, and CaOV3 resulted in a time- and dose-dependent synergistic effect on the inhibition of cell replication and induction of apoptosis.The combination treatment also showed enhancement of ROS production, causing DNA damage[45].Colorectal cancer cells resistant to 5-FUthat were treated with the combination of WA and 5-FU showed induction of ER stress, which eventually led to cell death.In these cells, in addition to inducing G2M phase arrest, WA worked by upregulating stress sensors including BiP,PERK, CHOP, ATF-4, and elF2a[46].
Hyperpolarization of the mitochondrial membrane is another mechanism of actionof WA.Targeting the mitochondria would influence mitochondrial metabolic activity, which could eventually lead to paraptosis.Cells treated with WA exhibited features (fusion of mitochondria and ER, expansion of vacuoles) that are indicative of paraptosis.Another notable observation in the treated cells was a decrease in the expression of Alix, anendogenous inhibitor of paraptosis[47].WA was shown to induce mitochondrial apoptosisand decrease the spread of cancer cells into surrounding tissues in drug-resistant breast cancer cells.It also led to the suppression of the Nf-kappaB/m-TOR signaling pathway[48].WA was shown to impair the proteolytic activity of lysosomes causing blockade of autophagic flux, a decrease in the substrates required for the TCA cycle and impaired oxidative phosphorylation, resulting in apoptosis in breast cancer cells[49].Similarly, another study concluded that the inhibition of proteasome activity and induction of impaired autophagy constituted the main mechanism associated with the antitumor effects of WA in breast cancer cells[50].WA was shown to induce G2/M cell cycle arrest in myelodysplasia and leukemia cells[51].Another study analyzed the effect of WA on pancreatic cancer cells, one of the most difficult forms of cancer to treat due to cell resistance to treatment.WA was shown to increase the number of autophagosomes, while simultaneously inhibiting the SNARE pathway, specifically the STX17 and SNAP29 receptors, which prevents autophagosome and lysosome fusion.WA was also shown to increase ER stress and inhibit proteasome activity, leading to an increase in ubiquitinated proteins in the cell.Pretreating the cells with TUDCA partially reduced WA-induced LC3-II accumulation, suggesting that ER stress precedes WA-induced autophagy.This study also investigated autophagosome formation by analyzing the encoding of the GFP-LC3-II protein with the lentivirus vector.Pancreatic cancer cellstreated with WA showed increased GFP-LC3-II in a dose-dependent manner, concluding that there was an increase in autophagosome formation[52].Another study sought to determine why WA was successful in inducing apoptosis in PC-3 and DU-145 but not in TIG-1 or LNCaP cells.In PC-3 and DU-145 cells, WA increased mRNA and protein levels of c-Fos.This did not occur in TIG-1 or LNCaP cells, which resulted in the ER stress response, eventually leading to cell death.A decrease in the expression of the anti-apoptotic proteinc-FLIP (L) was also observed in the study[53].When prostate cancer cells were treated with 3-AWA, an azido derivative from WA, LC3B-I was converted to LC3B-II, stimulating autophagy and eventually, apoptosis.Prostateapoptosis response-4 (PAWR or Par-4) is a protein closely associated with tumor-suppressing activity, and right before apoptosis, PAWR levels are elevated.When PAWR is overexpressed in a cell,it induces apoptosis by inhibiting the antiapoptotic protein BCL2 by binding to Wilms tumor 1 protein(WT1).This study found that because PAWR binds to WT1, it indirectly downregulates BCL2 expression and suppresses Beclin 1, an essential component in autophagy[54].WA has been shown to target oncogenic signaling pathways, specifically NF-κB, Akt, Stat3, and estrogen receptor-α (ER-α).In human cancer cells,these pathways are often hyperactive; however, WA has been shown to inhibit their activity.WA was shown to inhibit the NF-κB pathway by nuclear translocation of the p65 subunit of NF-κB and or downregulation of p6 in prostate cancer cells and soft tissue sarcoma cells.Suppression of ER-α by WA in breast cancer cells can lead to apoptosis.Cells treated with WA showed downregulated ER-α protein, leading to apoptosis[55].Upon exposure to WA, MDA-MB-231 and MCF-7 breast cancer cells underwent autophagy, which was confirmed by analysis of acidic vesicular organelles and cleavage and recruitment to autophagosomes of LC3-II[56].WA-induced autophagy was shown to be cytoprotective in hepatic cells, and by inhibiting its cytoprotective effects with chloroquine, tumor cells became more responsive to therapy[57].Table 3 summarizes the WA-induced autophagic response in different cancer models.
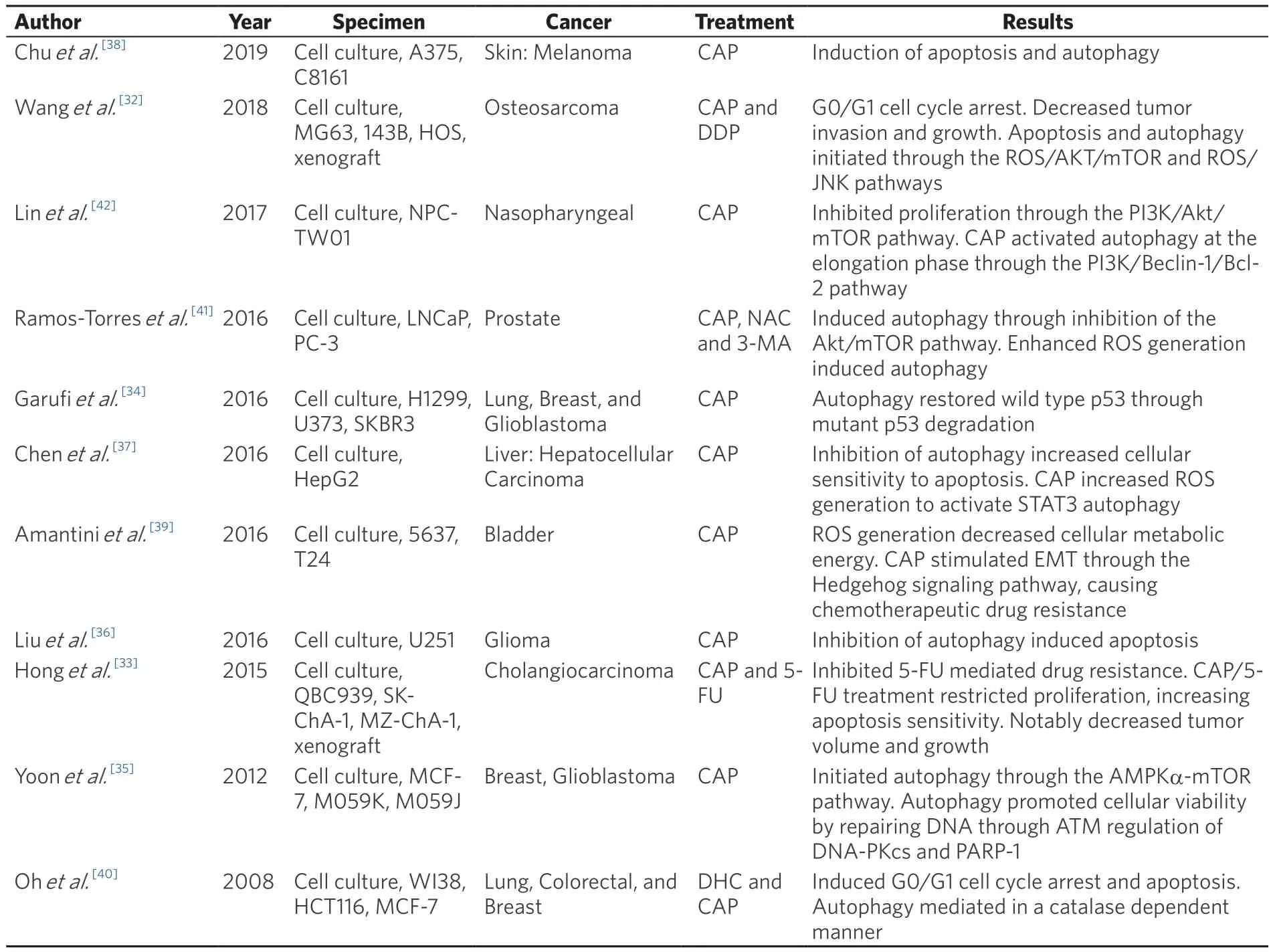
Table 2.Summary of autophagic response induced by Capsaicin in various cancer models
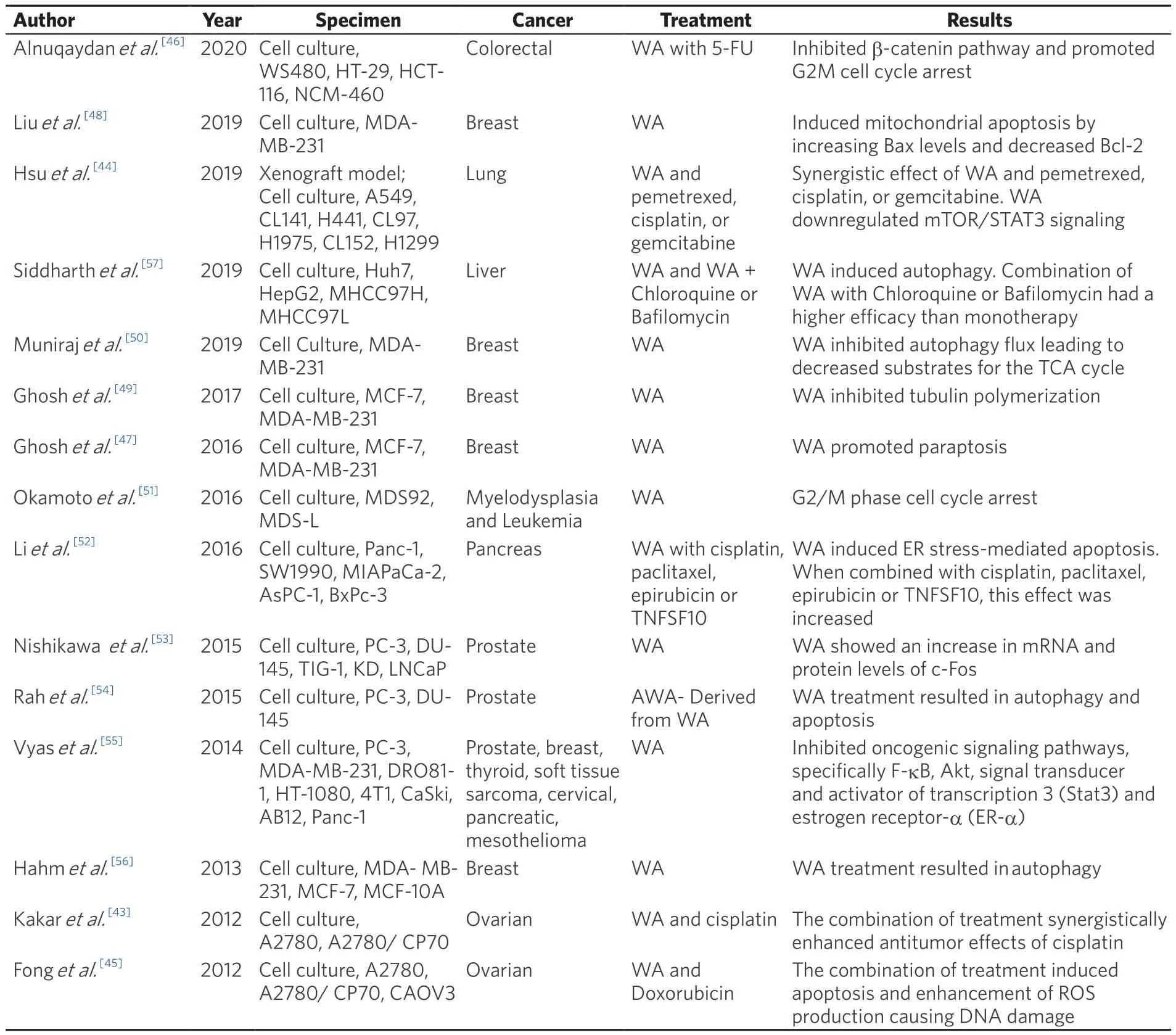
Table 3.Summary of autophagic response induced by Withaferin A in various cancer models
GENISTEIN
Genistein is a polyphenolic isoflavone compound derived from soy-based foods.The chemical structure of genistein is similar to that of estradiol, indicating its ability to bind to estrogen receptors.Genistein is soluble in polar organic solvents and has a lower solubility in water.Epidemiological studies have shown an inverse relation of soy-food intake and cancer development, especially in Asian countries where the consumption of soy-based foods is high compared to Western countries.Several meta-analyses have found that intake of soy foods and soy-based isoflavones is associated with prostate cancer reduction in men and breast cancer reduction in both pre- and postmenopausal women.Epidemiological evidence-guided research studies aimed at determining the molecular circuitry altered by genistein in cancer prevention by employing relevant cellular and animal models.Genistein is typically present in its glycosylated form during its biosynthesis in soybeans.After ingestion, deglycosylation occurs i n the small intestine, and the free form of genistein is absorbed in the body resulting in its various pharmacological effects[58].
Genistein’s antitumor effects have been extensively investigated in various cancer models including,ovarian, breast, prostate, and lung cancers and leukemia and melanoma[10,59].One of the earlier studies that investigated the antitumor properties of genistein in ovarian cancer cells reported the induction of apoptosis and autophagocytosis as potential mechanisms of cell death[60].In that study, A2780 ovarian cancer cells were transfected with a GFP-LC3 plasmid and subjected to glucose deprivation or different concentrations of genistein.Analysis of cells using fluorescence microscopy revealed recruitment and localization of LC3-II to autophagosomes, indicative of an autophagic response.Further experiments in the same study demonstrated that nutrient deprivation was an important part of autophagic cell death induced by genistein in ovarian cancer cells[60].A study focusing on elucidating the antitumor effects of genistein employing A549 lung adenocarcinoma cells concluded that genistein, by inhibition of autophagic flux, enhanced tumor necrosis factor-related apoptosis, inducing ligand (TRAIL)-induced cell death compared to genistein or TRAIL alone treated cells[61].Another study usingpancreatic cancer cells showed that genistein potentiated the antitumor effect of 5-FU by inducing apoptotic and autophagic cell death[62].That study used both cell culture and pancreatic tumor xenograft models and demonstrated that the combination of 5-FU and genistein had superior antitumor effects against the growth of pancreatic cancer cells when compared to the groups treated with 5-FU and genistein alone.Analysis of molecular mechanisms revealed increased expression of LC3-II, Beclin 1 and decreased levels of Bcl2, a key negative regulator of autophagy, which binds to Beclin1[61].In addition, the study also confirmed the induction of autophagic cell death in the group treated with both genistein and 5-FU by using chloroquine, a known inhibitor of autophagy[62].Prietschet al.[63]investigated the antiproliferative effects of genistein usingMCF-7 human breast cancer cells.The study demonstrated that the antitumor effect of genistein involved the generation of free radicals, increase in BAX/Bcl-2 ratio, and downregulation of survivin, which ultimately resulted in induction of apoptosis and autophagy.Even though the study did not demonstrate the exact role of autophagy, LC3-II immunostaining of the cells treated with genistein clearly revealed the induction of autophagy in breast cancer cells[63].Consumption of genistein (soy products or dietary supplements) was shown to have a differential effect in breast cancer cells with a varied ERα/ERβ ratio[64].Cells with a low ratio were shown to be more susceptible to genistein and increased effectiveness was seen in combination with tamoxifen-treated cells by increasing autophagic cell death[64].In contrast, a high ratio was shown to have counterproductive effect in anticancer treatment due to a decrease in ROS production, one of the main mechanisms of action of cisplatin (apoptosis) and tamoxifen-treated cells (autophagic cell death)[64].A study employing a preclinical model for estrogen receptor positive (ER+) breast cancer showed that lifelong genistein intake reduces the risk ofde novoand acquired tamoxifen resistance and recurrence of tumors.The findings from the study also showed that prepubertal and lifetime genistein consumption improved responsiveness to tamoxifen[65].The antitumor benefits associated with genistein intake was linked to its potential in boosting immunity against tumors, reducing unfolded protein response and pro-survival effects of autophagy[65].Phytochemicals’ ability to work synergistically and/or additively in the presence ofa different compound in lowering tumor burden has been reported for various cancers.Nakamuraet al.[66]showed synergistic antitumor effect by co-treatment with indole-3 carbinol and genistein against human colon cancer HT-29 cells.The antitumor effects were attributed to induction of apoptosis via inhibition of Akt phosphorylation and progression of autophagy.In a different study that useda combination of genistein and LC3 shRNA plasmid transfection, it was shown that the combination inhibited rapamycininduced autophagy and promoted apoptosis in human malignant neuroblastoma SK-N-BE2 and IMR-32 cells bothin vitroandin vivo[67].Malignant neuroblastoma is an extracranial solid tumor that usually occurs in children.Autophagy serves as a pro-survival mechanism in malignant neuroblastoma and usually deters the efficacy of conventional chemotherapeutic agents.The study revealed a decrease in expression of autophagy-related markers including LC3 expression and Beclin1 levels, and also modulated the expression of the apoptosis regulatory proteins Bax (pro-apoptotic) and Bcl-2 (anti-apoptotic) essentially to increase the Bax: Bcl-2 ratio to trigger the mitochondrial pathway of apoptosis[67].Most recently, a study that developed genistein-PEGylated silica hybrid nanoparticles against colorectal cancer showed that the genistein-based nanoparticles exerted improved antiproliferative effects by modulating antioxidant enzymatic activity and oxidative stress levels in human colon cancer HT29 cells.Furthermore, cells treated with the nanoparticles exhibited characteristics of autophagy and resulted in autophagic as well as apoptotic cell death without any side effects[68].
Radiotherapy continues to be an important treatment approach for several cancers.Genistein was shown to enhance the efficacy of radiation therapy in various cancer types.In a study employing non-small cell lung cancer (NSCLC) cells, genistein was shown to enhance cellradiosensitivity by inducing apoptotic and autophagic cell death.The findings revealed the role of genistein in enhancing radiosensitivity by increasing DNA damage, inhibiting cytoplasmic distribution of Bcl-xL and dissociation of Beclin1 from Bcl-xL to induce apoptosis and autophagy, respectively[69].
In addition to the beneficial effects discussed above, either alone, with estrogen receptor modulator(tamoxifen) or radiotherapy, genistein has also been found to increase the potency of several traditional cytotoxic chemotherapeutic agents.For example, a recent study showed that genistein potentiated arsenic trioxide (As2O3) treatment of acute promyelocytic leukemia induced apoptosis and autophagy with the combination treatment.The study revealed that genistein in combination with As2O3was able to increase the total release of ROS andalso the expression ratio of LC3-II/LC3-I in NB4 cells[70].In a similar study,both cisplatin-sensitive A2780 and cisplatin-resistant ovarian cancer cells were treated with genistein alone.The findings revealed that genistein inhibited AKt kinase, which is an oncogenic kinase that plays an important part in glucose uptake.The resulting low glucose intracellular environment induced autophagy,which was evident by an increase inLC3-II levels[71].In a different study, genistein was used to treat uterine leiomyoma (UtLM) cells, in which MAP1LC3A was used as a marker to determine an autophagic response.The findings revealed induction of autophagy and subsequent apoptotic cell death by genistein in UtLM cells 72 hours after treatment[72].Table 4 summarizes the genistein-induced autophagic response in different cancer models.
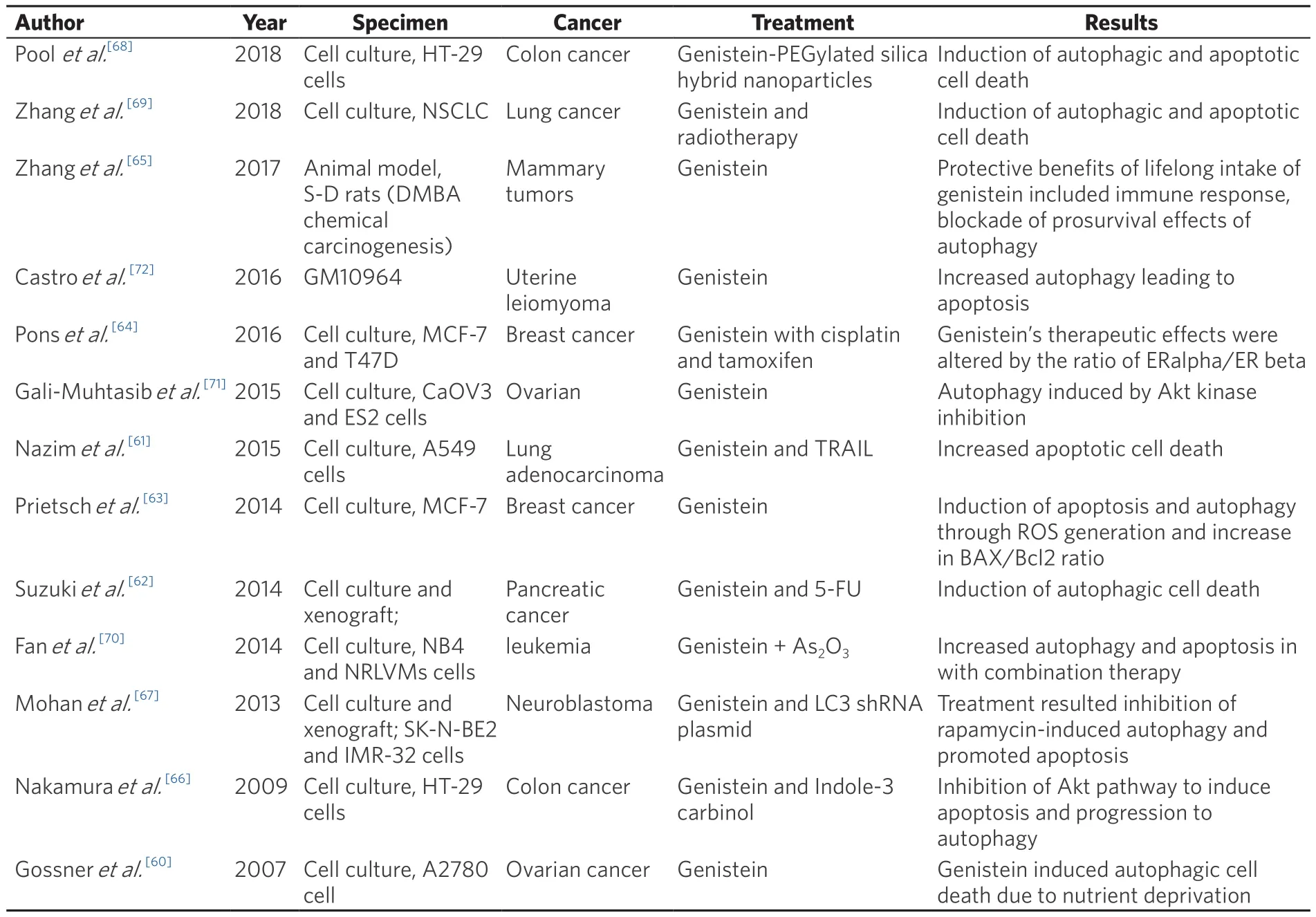
Table 4.Summary of autophagic response induced by Genistein in various cancer models
Other major phytochemicals known to induce autophagic response
In addition to the four phytochemicals discussed in the current review, several other phytochemicalshave been found to induce autophagic response in various cancer models.For example, curcumin[11], a polyphenolic compound present in turmeric, was shown to induce autophagic cell death in malignant glioma cells by inhibiting the Akt/mTOR/p70S6k pathway and activating the ERK1/ERK2 pathway[73,74].A most recent study showed that combination of mTORC1/2 inhibitor with curcumin induced autophagymediated cell death by reducing the expression of Rictor and Akt in renal carcinoma,in vitroand in a xenograft model[75].Similarly, resveratrol[12], another major polyphenolic compound present in various natural products including grapes, is well known for its antitumor effects in various cancer models, bothin vitroandin vivo.For example, resveratrol was shown to induce autophagy-mediated cell death in ovarian cancer, breast cancer and leukemia cells[76-78].It was shown that genetic or pharmacological modulation of autophagic proteins could play a major role in resveratrol-mediated cell death[79].A recent study with ovarian cancer cells showed that treatment of the cells with resveratrol resulted in growth suppression through epigenetic modulation of genes and microRNAs that are associated with the invasive phenotype[80].Honokiol, a phenolic compound from the magnolia treewas studied for its protective effects against chronic ailments including cancer.Numerous studies identified the potential of Honokiol to induce autophagic cell death in various cancer models[81-84].For example, a recent study showed that generation of intracellular ROS, activation of ERKs and upregulation of Atg7 were essential to Honokiol-mediated autophagic cell death in OS[85].
A schematic diagram focusing on the mechanism by which different phytochemicals induce autophagy is presented in Figure 1.
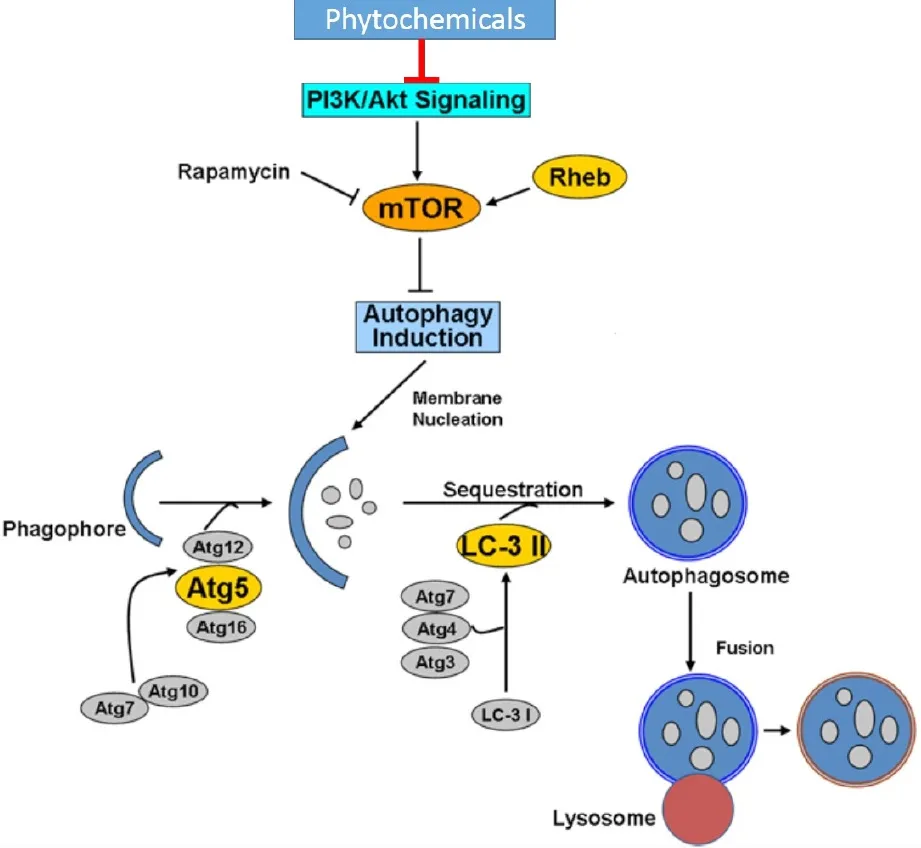
Figure 1.Schematic diagram representing the mechanism by which phytochemicals inhibit PI3k/mTOR pathway to induce autophagy.Induction of autophagy ensues upon negative regulation of mTOR that leads to formation of a phagophore and subsequent sequestration of cellular constituents in the presence of important autophagy proteins (Atg proteins) and cleaved LC3-II.Fusion of the autophagosome with lysosome results in lysis and generation of energy that is typically expended for cell survival or cell death (Type II).mTOR:mammalian target of rapamycin
Limitations of phytochemicals
A large body of evidence suggests the inverse association of consumption dietary compounds rich in phytochemicals discussed in this review and the development of various malignancies including cancer.Despite the promising preclinical evidence, clinical development of these phytochemicals has faced severe roadblocks owing to their pharmacokinetic profile.The genetic profile of individuals is known to play a major role in the tissue distribution and elimination of various phytochemicals.For example, in a randomized study, glutathione S-transferase (GSTM-1)-positive individuals who consumed broccoli,a dietary source of sulforaphane (isothiocyanate) were shown to excrete sulforaphane metabolites at ahighest rate.Higher and rapid elimination of phytochemicals/metabolites indicates decreased biological benefits at the tissue level in the individuals who are positive for the enzyme involved in its metabolism[86].Similarly, studies identified that isothiocyanates (especially sulforaphane) are not effective as therapeutic agents against chronic conditions that are known to be associated with oxidative stress[86].Pharmacokinetic studies in preclinical models and human trials have shown that phytochemicals are avidly metabolized in the body, which could limit theavailability of these compounds ortheir associated active metabolites at the target organs distinct from the site of absorption.The therapeutic benefits of phytochemicals could only be realized through repeated administration, which would eventually produce systemic concentrations at the target sites.In addition, several studies including those discussed in the current review are exploring the possibility of increasing the bioavailability of these compounds through pharmaceutical modifications.Another important limitation of the administration of phytochemicals is their ability to cause the generation of ROS, which plays a major role in their pharmacological effects.It is important to limit the oxidative damage caused by ROS in the body.If uncontrolled, ROS may lead to the development of various chronic diseases and contribute to toxic side effects.It is because of these limitations that phytochemicals are not having much success in clinical trials and hence are being investigated extensively to overcome these obstacles.
FUTURE PERSPECTIVES AND CONCLUDING REMARKS
The relevance of autophagy in chronic diseases has been well established and shown to have a major role in the development of these ailments, including cancer.Autophagic response in cancer development is both tumor-suppressive and tumor promoting depending on such factors as cancer type, energy status and metabolic processes within a cell.Moreover, the regulation of autophagy by phytochemicals in cancer cells also depends on epigenetic mechanisms that modulate the expression of key proteins involved in its induction[87].The overall chemopreventive effects of various phytochemicals[88], including those discussed in the current review, were also attributed to their ability in regulating microRNA levels.Understanding the molecular mechanisms by which autophagy playsa role in cancer stem cells is critical in developing novel therapies[89].Cancer cells can stimulate the process of autophagy for accessing nutrients in their microenvironment for their growth and proliferation, and hence, understanding the role of autophagy in its microenvironment could result in radical approaches for a better therapeutic outcome.In this review,we discussed four major phytochemicals that are abundantly present in various dietary sources and consumed by different cultures across the world.These phytochemicals are shown to induce autophagic response in different cancer models.The autophagic response evoked by phytochemicals not only can mitigate resistance to conventional therapiesbut also serve as a type (type II) of cell death.The induction of autophagy by different phytochemicals depends on various factors, including the type of cancer.For example, genistein was shown to potentiate the antitumor effect of 5-FU by inducing apoptotic and autophagic cell death.Similarly, CAP and cisplatin combination induced autophagy that served as a prosurvival mechanism in OS cells, which when inhibited by bafilomycin resulted in upregulation of apoptotic genes.WA-induced autophagy was shown to be cytoprotective in hepatocellular carcinoma, which was abrogated in the presence of autophagy inhibitors, increasing the efficacy of combination therapy.The autophagic response induced by PEITC increased overall cell death in prostate cancer cells, while the response preserved the metastatic potential of lung cancer cells, which was abrogated in the presence of chloroquine.In addition, PEITC’s ability to modulate the expression of miR-194, MMP2, and MMP9 proteins was vital in the induction of autophagy and prevention of metastatic potential in prostate cancer cells.
A continued progress in understanding the pharmacology of these phytochemicals and advances made to overcome the challenges associated with their pharmacokinetic profile have enabled their commercialization.The market for various phytochemicals continues to grow as more and more people are looking for alternate approaches in promoting their health and preventing disease development.Several phytochemicals are marketed as dietary supplements and are widely available in various forms.In conclusion, the utility of phytochemicals as therapeutic agents when used either alone or in combination with other conventional therapies can only be achieved after addressing the pitfalls and understanding the disease progression.
DECLARATIONS
Acknowledgments
The authors would like to acknowledge the faculty development funds awarded to Bommareddy A by Wilkes University and to thank Dr.Paul S.Adams and the mentoring task force at Wilkes University for providing summer research stipends to undergraduate students.
Authors’ contributions
Responsible for compiling information along with the tables related to PEITC, capsaicin, withaferin A and genistein A: Wenner C, Stewart R, McCabe A, Pepe J, Oberlin J
Then I had an unexpected visit from the person I dreaded, most to see -- the mother of the college classmate who died. Years ago , she said, I found it in my heart, through prayer, to forgive you. Betty forgave you. So did your friends and employers. She paused, and then said sternly,, You are the one person who hasn t forgiven Tom Anderson. Who do you think you are to stand out against, the people of this town and the Lord Almighty5? I looked into her eyes and found there a kind of permission to be the person I might have been if her boy had lived. For the first time in my adult life I felt worthy to love and be loved.
Helped with the figures and future perspectives section: VanWert A
Conceived the idea and compiled information related to introduction, limitations to phytochemicals and future perspectives & conclusions and was responsible for the preparation of the final article: Bommareddy A
Availability of data and materials
Not applicable.
Financial support and sponsorship
Not applicable.
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2020.
 Journal of Cancer Metastasis and Treatment2020年9期
Journal of Cancer Metastasis and Treatment2020年9期
- Journal of Cancer Metastasis and Treatment的其它文章
- Overview of genetic signaling pathway interactions within cutaneous malignancies
- Liquid biopsy in endometrial cancer
- Epithelial-mesenchymal transition: a hallmark in pancreatic cancer stem cell migration, metastasis formation, and drug resistance
- The good and bad sides of exosomes: premetastatic niche formation, cancer biomarker and therapy carriers
- Extracellular RNAs as potential biomarkers for cancer
- Exploiting the relevance of CA 19-9 in pancreatic cancer