
生物催化合成手性胺的研究进展
2020-07-15 01:56刘婉颐邓国忠崔宝东钟永科
合成化学 2020年6期
刘婉颐, 邓国忠*, 崔宝东, 钟永科
(1. 遵义医科大学 a. 药学院; b. 贵州省生物催化与手性合成重点实验室, 贵州 遵义 563000)
手性胺类化合物是多种药物活性分子的重要结构单元,在生物医药和农用化学品中扮演着重要的作用。据统计,许多手性药物分子都含有手性胺的分子骨架,如卡巴拉汀(1a)、达泊西汀(2a)、加雷沙星(3a)等(Chart 1)[1]。此外,手性胺还可以作为重要的手性配体[2]。在传统的合成方法中,手性胺的合成主要依赖于烯胺还原、酰胺还原、亚胺还原及不对称转移氢化等[3],常用的催化剂以有机小分子催化剂和金属催化剂为主,小分子催化剂如手性磷酸、手性双烯-硼烷,金属催化剂钯、钛、铑、钴等[4]。
Scheme 1
近年来,生物催化因具有反应条件温和、选择性高和环境友好等特点,不断应用于手性胺类药物分子及其中间体的合成应用之中[5-6]。自然界提供了物种丰富的生物酶库,为手性药物分子的合成应用和工业生产提供了物质基础。随着DNA测序、DNA重组和酶蛋白的定向进化等生物技术的快速发展,越来越多的生物催化剂在化学反应中有了更优异的表现[7],而生物催化合成手性胺的方法也不断涌现。本文综述了近年来不同生物酶在手性胺酶法合成中的应用。
Scheme 2
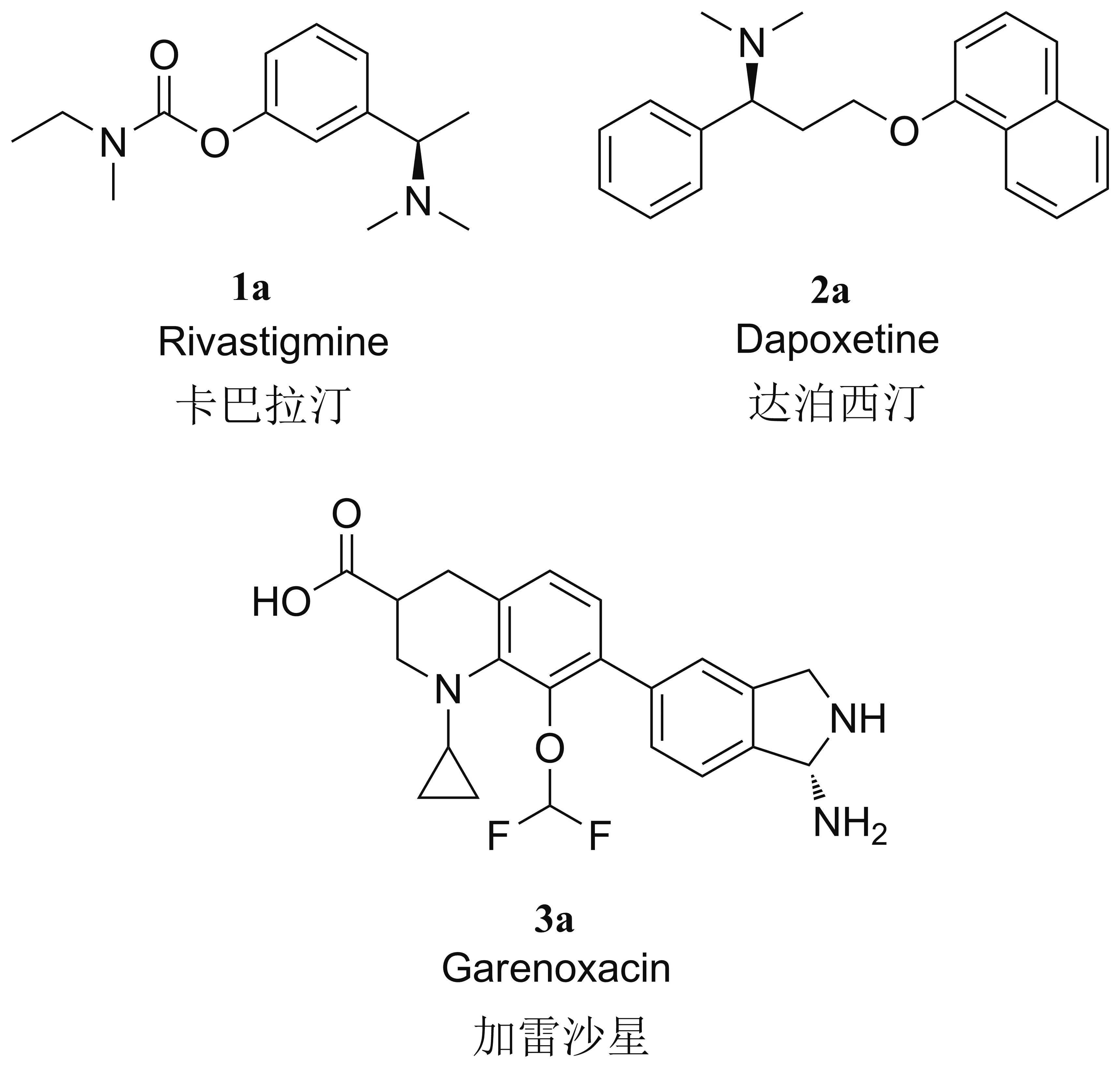
Chart 1
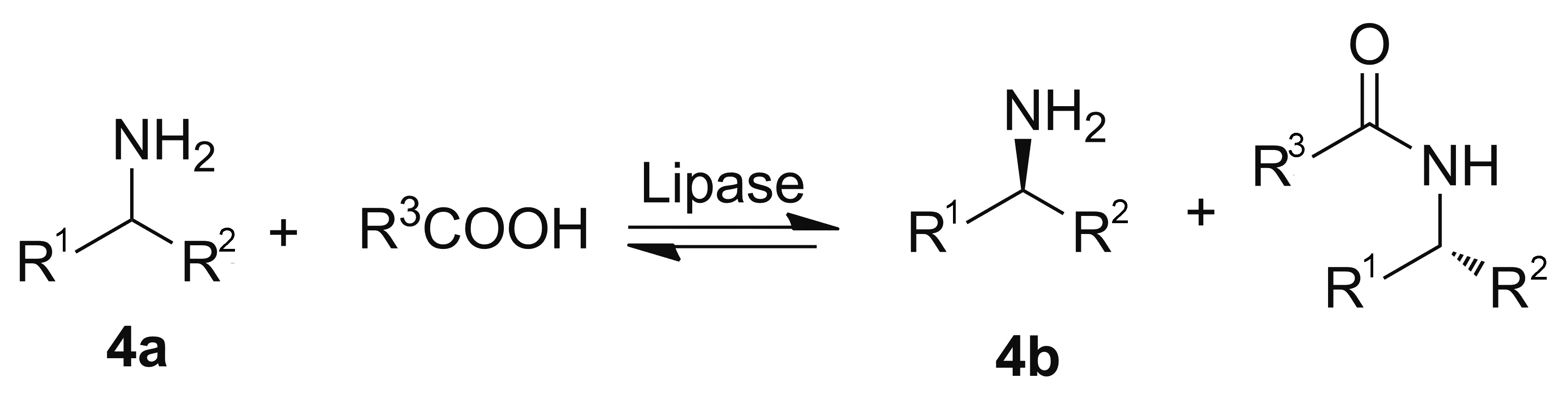
脂肪酶(Lipase)来源于羧基酯水解酶家族,能够催化酯键和酰胺键的断裂或形成。脂肪酶具有多种催化活性,不仅可以催化拆分外消旋醇类、酸类、胺类和酯类,而且可以催化酯水解、胺解、酯交换等反应[10]。脂肪酶催化手性胺的合成策略一般是以外消旋的胺(4a)合成外消旋的酰胺或酯,再进行动力学拆分得到手性胺(4b, Scheme 1)[5]。
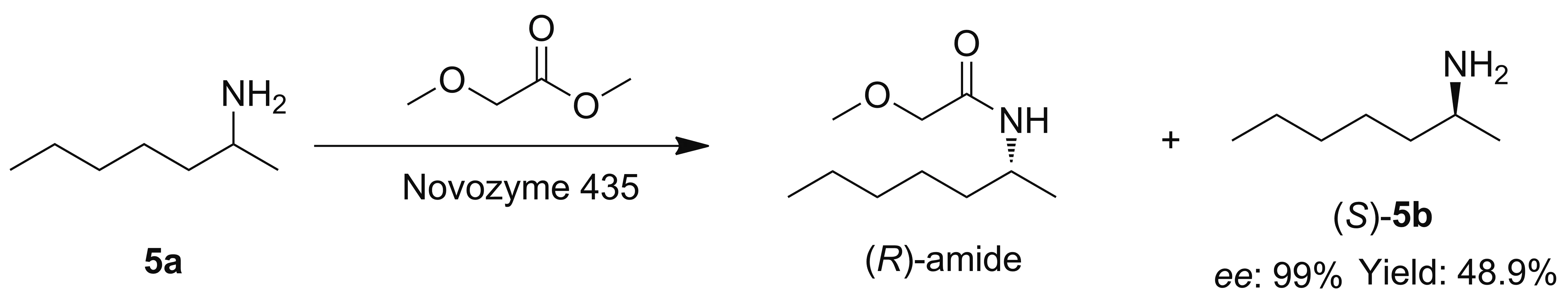
1996年,Kohls[11]课题组首次报道了以脂肪酶作为生物催化剂对手性胺进行动力学拆分。90年代初期,脂肪酶在工业上开始得到广泛的应用,拜耳公司利用lipase B对α-甲基苄胺进行了动力学拆分。2010年,SUN[13]课题组利用脂肪酶Novozyme 435对2-庚胺(5a)进行动力学拆分,得到了99%的ee值和收率48.9%的(S)-5b(Scheme 2)。
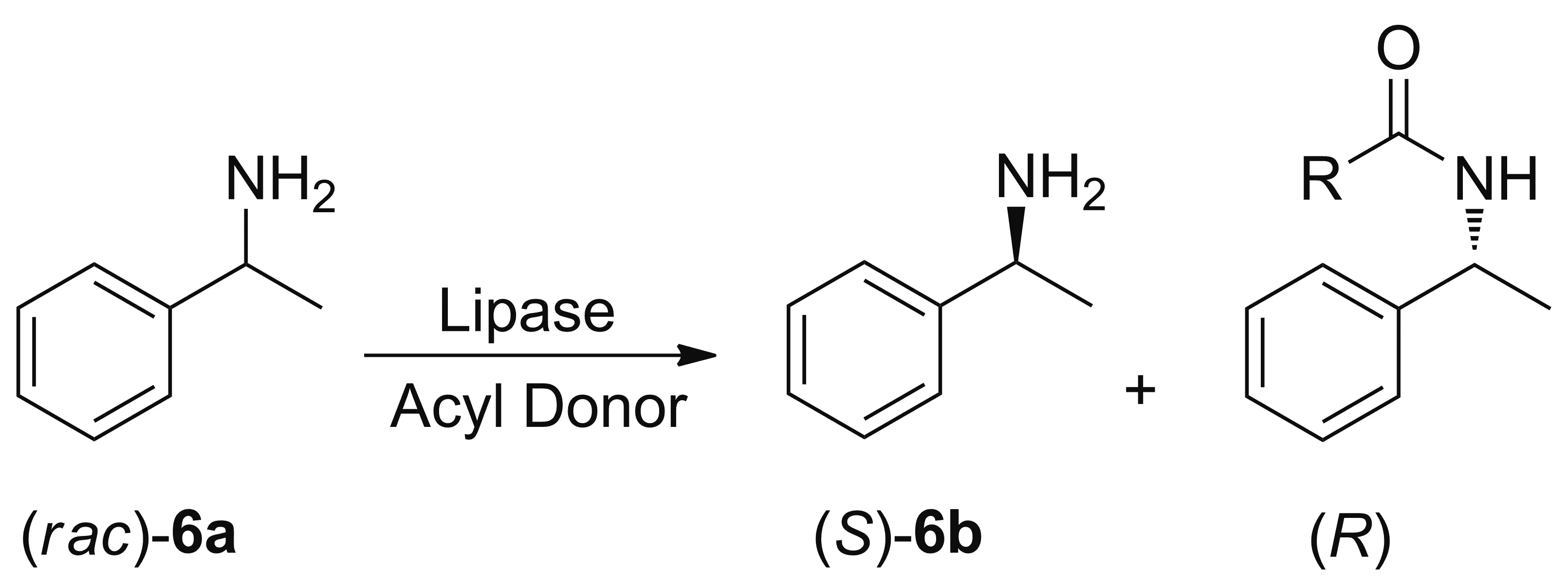
2011年,Munoz[14]课题组利用脂肪酶为催化剂合成高对映选择性的苯乙胺。2013年De Miranda[15]课题组报道了使用丙烯酸树脂固定的Novozyme 435脂肪酶作为生物催化剂,对1-苯乙胺(6a)进行酯化拆分,得到(S)-构型的苯乙胺(6b, Scheme 3),催化底物浓度高达900 mM,其固定化酶可以进行9次有效循环仍保持较高催化活性;发现在不同酰基供体下,该反应的转化率为31%~57%不等。温和条件下,当以2-甲氧基乙酸甲酯或乙酸乙酯作为酰基供体时,该反应体现出优异的对映选择性,E值可达200以上,但随着反应温度的升高E值呈现下降的趋势。
Scheme 3
Scheme 4
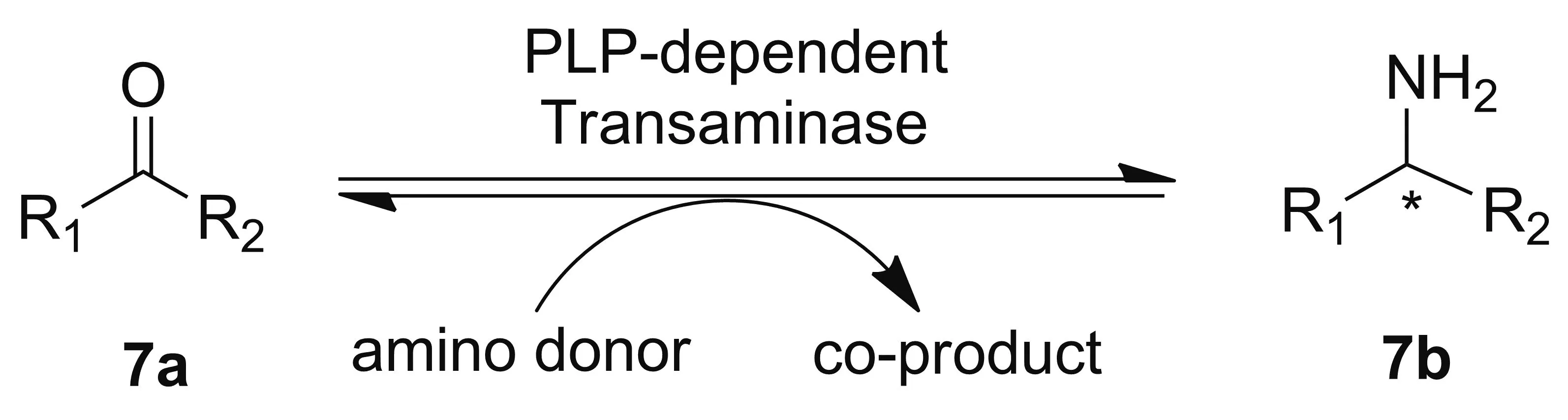
转氨酶(Transaminases, TAs)是一类5′-磷酸吡哆醛(PLP)依赖性转移酶,能够可逆催化酮基与氨基间的立体选择性转移,其普遍存在于植物、微生物以及动物的心肌、脑、肝、肾等组织中[5]。转氨酶根据氨基转移位置的不同,大致可分为α-转氨酶和ω-转氨酶两大类,当催化的反应底物或产物中含有α-氨基酸时称之为α-转氨酶,含有ω氨基酸则为ω-转氨酶[16]。α-转氨酶的产物较为单一,通常仅为α-氨基酸,而ω-转氨酶因其可以催化酮、醛等不对称氨基转移从而在医药合成中得到更加广泛的应用[17]。转氨酶在催化反应过程中不需要昂贵的辅因子参与反应,可将酮或醛(7a)与氨基供体反应生成手性胺类化合物(7b,Scheme 4),但其催化活性易受到底物抑制和产物抑制,并且底物范围相对较窄[9]。可逆反应的存在是将酮类底物转化为手性胺的主要障碍,而去除副产物丙酮酸可改善这种不利的热力学不平衡[18]。转氨酶用于手性胺合成的方法大致分为三类:(1)外消旋体的动力学拆分;(2)前手性酮的不对称转化;(3)外消旋底物的转化[19](图1)。在90年代初期,STIRLING DAVID等[20]就报道了(R)-构型和(S)-构型的转氨酶,能够将酮类底物氨基化为手性苯甲胺,收率均在90%以上。

Scheme 5
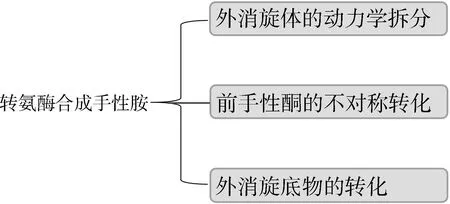
图 1 转氨酶合成手性胺的策略
2010年,Savile等[21]以转氨酶为生物催化剂,取代铑催化烯胺进行不对称加氢合成手性胺,用于工业化生产抗糖尿病药物西他列汀(8c),这是生物催化在工业生产上的重大突破,也标志着生物催化在合成手性胺类药物活性分子中的合成应用(Scheme 5)。
2015年,Chen课题组克隆表达了一种来自热脱硝杆菌的(S)-构型选择性的ω-转氨酶TPTAgth,在pH 9.0和65 ℃下仍可以保持高活性,表现出良好的耐热性和强碱环境耐受性,且该酶对酮糖具有良好的催化活性。
2016年,PAVLIDIS等在取代基空间位阻较大的结构方面得到了较大进展,能够将体积较大的酮转化为相应的手性胺(9a~14a),其收率最高可达82%,ee值可达99%以上,表现出优秀的对映选择性(Chart 2)。

Chart 2
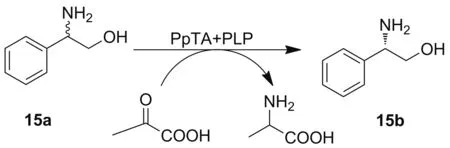
2016年,Wu课题组[24]发现了4个来自假单胞菌的(R)-构型选择性ω-TAs,其中PpspuC通过动力学拆分外消旋2-苯甘氨醇(15a)得到ee值高于99%的(S)-2-苯甘氨醇(15b)(Scheme 6)。2019年该课题组克隆表达了来自节杆菌属的(R)-构型选择性ω-TAs(MVTA),可以对大体积的β-氨基醇进行动力学拆分,其产物的ee值大于99%[25]。
Scheme 6

Scheme 7
在之前的报道中,绝大部分转氨酶属于(S)-构型选择,报道的(R)-构型选择的转氨酶较少,只有一种来自节杆菌属的转氨酶被设计来转化大基团的酮。2016年,Guan课题组[28]报道了对来自节杆菌属的(R)-转氨酶的晶体结构分析,为此类转氨酶的结构修饰改造、定向进化提供了结构基础和理论依据,并为新型手性胺类化合物合成途径的设计奠定了基础。

Chart 3
Scheme 8
单胺氧化酶(Monoamine oxidases, MAOs)属于黄素腺嘌呤核苷酸(FAD)依赖的胺氧化还原酶类,催化由氧驱动的胺向相应的亚胺转化,并生成副产物过氧化氢。由于单胺氧化酶的高立体选择性识别,通常只选择将单一构型的对映异构体进行转化,从而将消旋的手性胺去消旋化而获得单一构型的产物。1995年,来自Asoergillusniger的单胺氧化酶首次被发现并命名为MAO-N[30]。2002年,Alexeeva课题组[31]报道了利用MAO-N为生物催化剂,通过MAO-N催化的氧化拆分反应制备手性胺的新策略。
2013年,Ghislieri课题组[32]利用MAO-N为生物催化剂,对四氢-β-咔啉(16a)结构进行了去消旋化反应,并得到ee值为99%和93%的单一构型产物(16b, Scheme 7)。该课题组进一步对MAO-N进行定向进化,扩大了含有芳基取代的手性胺底物谱(17a~29a, Chart 3)[7]。2018年,该课题组对单胺氧化酶可催化的底物范围进行了总结,其中突变体D5、 D9、 D11具有较高的活性,其底物范围覆盖了α-取代的甲基苯胺、联苯胺和1,2,3,4-四氢喹啉等不同结构的伯胺、仲胺、叔胺化合物。
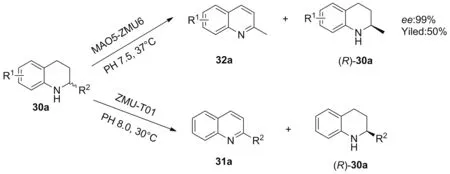
2016年,本课题组发现蒙氏假单胞菌ZMU-T01的整细胞对四氢喹啉(30a)类化合物具有氧化拆分活性,以此菌株整细胞作为生物催化剂能对2-取代-1,2,3,4-四氢喹啉(31a)进行氧化拆分,并以最高99%的ee值和50%的收率获得(R)-构型产物。该菌株整细胞对2-取代-1,2,3,4-四氢喹啉类底物普适性较好,但对于苯环含有取代基的底物适应性表现不佳[34]。2018年,本课题组从蒙氏假单胞菌ZMU-T01中克隆表达了一株新的单胺氧化酶MAO5,构建相应基因工程菌,通过整细胞生物催化反应实现苯环取代的2-甲基-1,2,3,4-四氢喹啉(32a)类化合物的高对映选择性氧化拆分,对多种底物表现出优异的立体选择性,ee值可达99%及以上(Scheme 8)[35]。
2016年,Li课题组利用来自短杆菌IH-35A的环己胺氧化酶(Cyclohexylamine oxidase, CHAO)为生物催化剂,对外消旋胺进行动态动力学拆分去消旋。野生型的CHAO对肿胺和叔胺均表现为低活性或无活性,但通过对CHAO进行定向进化,使其对2-取代-1,2,3,4-四氢喹啉类化合物可进行有效的动态动力学拆分,并得到了高达99%的ee值和76%的收率。他们对CHAO进一步研究并对其晶体结构进行分析,通过定向进化,拓展该酶的底物结合口袋,成功筛选到突变体,可以有效催化2-位取代基为大基团的底物,并具有较高的对映选择性[37]。2018年,朱敦明课题组与Reetz教授课题组合作共同对MAO-N进行改造,使其对1,2,3,4-四氢-1-取代异喹啉、2-苯基吡咯啉等(33a~37a)底物都有较好的底物适应性,并且表现出较好的对映选择性和催化活性[38]。
亚胺还原酶(Imine reductase, IRED)是一种依赖NAD(P)H的氧化还原酶,可以催化前手性亚胺不对称还原为相应的胺,具有高度立体选择性[39]。除单胺氧化酶可用于广泛结构的手性胺的合成外,其余大多数生物催化剂只能产生一级胺,不能直接生成二级胺或三级胺,转氨酶和氨脱氢酶都仅限于将氨基转移至羰基上,对于手性仲胺和叔胺的合成是难以实现的。而亚胺还原酶可以通过不对称氢化还原为这些手性胺化合物提供新的合成方法。亚胺还原酶虽然能催化反应生成手性胺,但其对映选择性较低,并需要大量的生物催化剂加入[42-43]。2010年,Mitsukura等[44]报道了来自链霉菌属的亚胺还原酶GF3587能够将2-甲基吡咯啉(38a)还原为相应的(R)-2-甲基吡咯烷(38b)。 2011年,该课题组将另一种亚胺还原酶GF3546进行纯化表征和表达,能够催化生成(S)-2-甲基吡咯烷(38c, Scheme 9),其催化活性为野生型的两倍[45]。
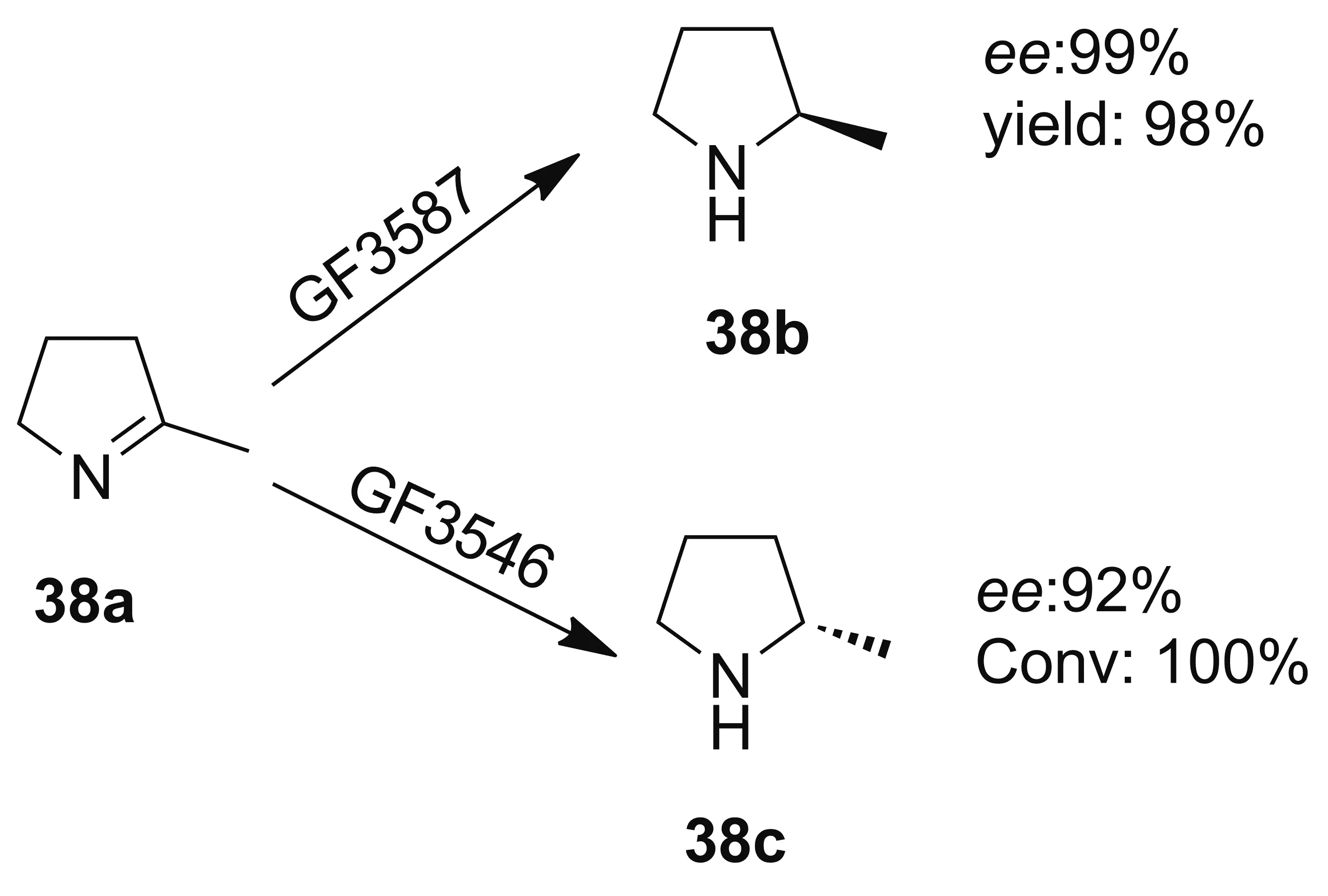
Scheme 9
2013年,Leipold课题组[46]将亚胺还原酶GF3546过表达于大肠杆菌宿主中,其催化底物普适性得到提升,可以对映选择性还原一系列不同的亚胺,包括2-取代环亚胺,二氢异喹啉等。2015年,该课题组将GF3587过度表达于大肠杆菌中,其催化活性得到了较大提升[40]。同年,联合亚胺还原酶((R)-IRED)和单胺氧化酶(MAO-N)对外消旋杂环胺(39a)进行动态动力学拆分,得到ee值>99%手性的杂环胺(39c, Scheme 10)[47]。 MAO和IRED共同作用时可用于手性胺的动态动力学拆分,而这种双酶级联催化反应体系避免了MAO在催化胺类化合物动态动力学拆分过程中化学还原剂的使用,如氨硼烷。
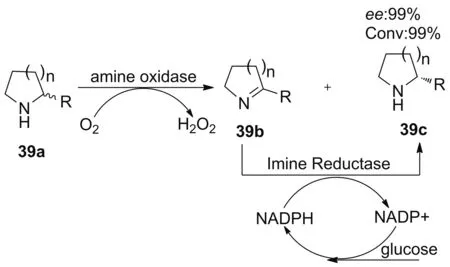
Scheme 10

Scheme 11
2018年,Velikogne课题组[39]将亚胺还原酶与醇脱氢酶进行共表达后得的菌株,通过醇脱氢酶在NADP+作为辅因子下可氧化脱氢2-丙醇生成丙酮,并得到亚胺还原酶完成亚胺还原反应所需的NADPH,从而达到辅因子循环。在不添加其他辅因子的条件下,底物浓度可提升至100 mM,为克级制备光学纯手性胺提供了一种绿色、高效且经济的方法。
Scheme 12

Scheme 13

Scheme 14

Scheme 15
胺脱氢酶(Amine dehydrogenase, AmDH)是一种能够催化酮的不对称还原胺化反应的生物酶,且副产物仅有水。早在1968年Eady等[48]就对甲胺脱氢酶进行了纯化表征,但其不能依赖NAD+或NADP+为电子受体,并且仅能氧化脂肪族的单胺、二胺、组胺,不能氧化仲胺、叔胺和季胺盐。2000年,Itoh等[49]对来自链霉菌中的胺脱氢酶进行分离纯化表征,并构建了它的催化底物范围,不仅能催化胺的可逆氧化脱胺反应,还能催化氨基醇和氨基酸的可逆脱胺反应,但催化效率和对映选择性都不高;但此次构建的氨脱氢酶基因工程菌可以依赖NAD+或NADP+进行电子传递,而之前所报道的此类酶均需要人工电子受体。2012年,Abrahamson[50]课题组对来自芽孢杆菌的亮氨酸脱氢酶进行定向进化,开发了一种新的氨脱氢酶,具有较高的催化活性,能催化4-甲基-2-氧代戊酸(41a)和甲基异丁基酮(42a)生成相应的手性胺(41b,42b, Scheme 11),ee值达到99%。
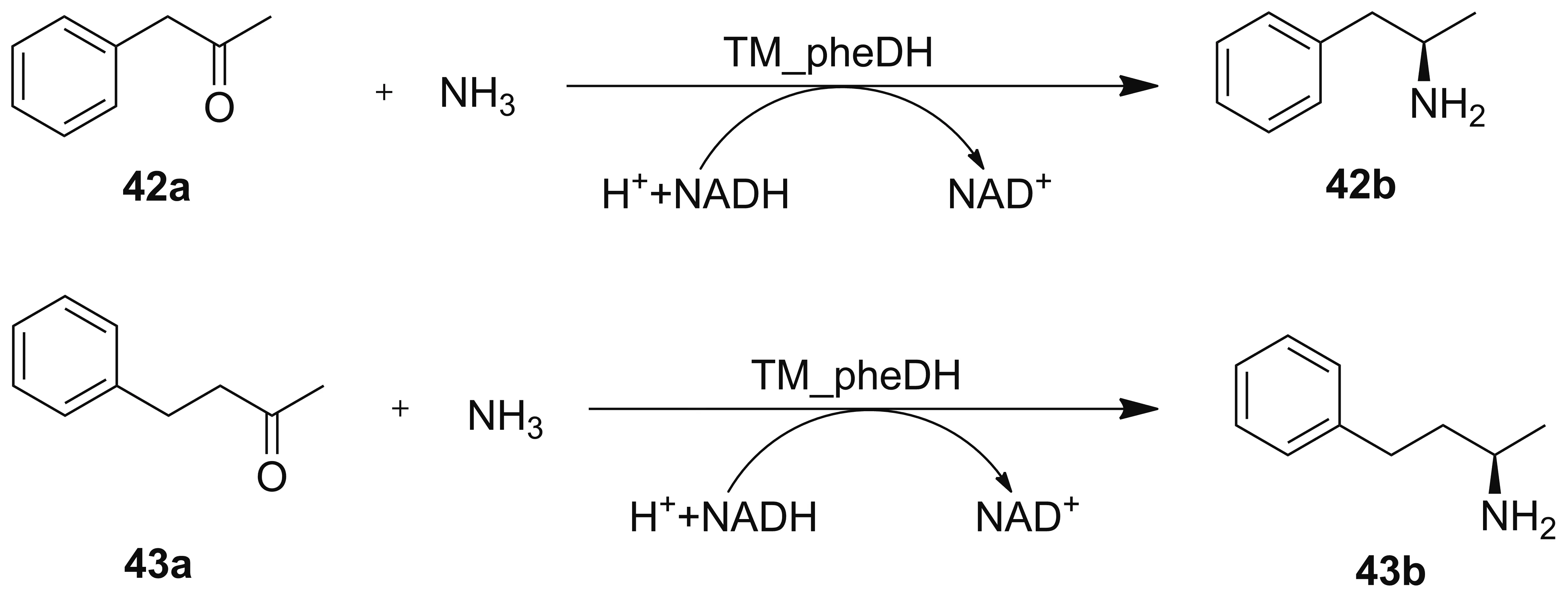
2015年,Ye课题组[51]发现了来自红球菌属的苯丙胺酸脱氢酶通过定向进化得到的三突变体K66Q/S149G/N262C(TM_PheDH)是具有高度对映选择性的生物催化剂,能选择性地还原苯基丙酮(42a)和4-苯基-2-丁酮(43a),得到ee值高达98%的(R)-安非他明(42b)和(R)-1-甲基-3-苯基丙胺(43b, Scheme 12)。

Scheme 16
2016年,Abrahamson课题组[50]开发了3个胺脱氢酶与甲酸脱氢酶协同作用,可对各类芳香族和脂肪族的酮类及醛类进行还原胺化,其催化的底物浓度高达50 mM,同时达到99%的ee值,利用此双酶体系进行还原胺化,仅生成无机碳酸盐为唯一副产物。并在他的研究中证明了单一氨脱氢酶不能接受不同类型的底物,如Ch1-AmDH对脂肪族酮和苯乙酮(44a)有较高的活性,Rs-PhAmDH对苯丙酮衍生物(45a)及空间位阻较大的酮有较高的活性(Scheme 13)。
2017年,Ye课题组[51]利用苯丙氨酸脱氢酶对苯氧基-2-丁酮(46a)进行还原胺化得到(R)-1-苯氧基-2-丙胺(46b, Scheme14),其底物浓度可以达400 mM,产物ee值>99%,转化率为96%。
细胞色素P450单加氧酶(Cytochrome P450 monooxygenases)是自然界用途最广泛的生物催化剂,具有催化反应类型多样性和底物多样性[52]。可以催化多种底物的区域选择性和立体选择性氧化反应,并参与多种天然产物的生物合成[53]。
早在1985年就曾有利用P450酶催化氮原子的功能化转移报道,这是P450在胺类化合物合成中较早的应用[54]。2013年,Mcintosh课题组[53]报道了用于C—H胺化的高活性酶催化剂,对P450BM3的T268A和C400S残基突变后获得P450BM3变种酶P411,其对C—H胺化的催化活性得到了巨大提升,能催化2,4,6-三乙基苯磺酰叠氮(47a, Scheme 15)生成环状手性胺(47b)(Scheme 15),ee值为87%,分离收率69%。
2016年,Prier课题组[55]继续以P411突变体为生物催化剂,利用σ重排进行烯丙基胺的不对称酶法合成。2019年,该课题组继续以P411为模板进行突变,构建了一个可以用于一级(48a),二级(49a)和三级(50a, Scheme 16)C—H不对称胺化的酶库,并在吸电子基和给电子基以及空间位阻的影响下都具有良好的底物普适性[56]。
手性胺作为药物合成中的重要骨架分子,在新药研发和天然药物化学结构中都具有重要的意义。近些年来,随着分子生物学技术的发展,使得生物酶定向进化、理性设计等技术愈发成熟。可以根据不同的化学反应设计所需的生物酶,尤其在区域选择性、对映选择性控制以及不活泼位点的活化方面取得显著提升。现阶段所发现的脂肪酶、转氨酶、单胺氧化酶、亚胺还原酶、胺脱氢酶以及P450酶已经能在不同类型的手性胺合成中具有一定的特点和优势。但目前仍然存在新酶发现不足,底物普适性不高、热稳定及有机溶剂耐受性有限等问题,这些问题也是指导手性胺类药物分子重要中间体生物催化合成发展的重要方向。
猜你喜欢
分子催化(2022年1期)2022-11-02
科学导报(2022年41期)2022-07-13
昆明医科大学学报(2021年5期)2021-07-22
湖南大学学报·自然科学版(2020年2期)2020-04-17
科技创新导报(2019年14期)2019-10-20
宇航学报(2018年10期)2018-11-08
上海航天(2018年3期)2018-06-25
飞控与探测(2018年1期)2018-04-18
江苏农业科学(2017年20期)2017-11-30
吉林农业·下半月(2016年11期)2017-01-09
