
孑遗濒危植物矮扁桃叶绿体全基因组特征分析及亲缘关系鉴定
2020-07-14 01:00:16孟庆瑶段义忠
植物研究 2020年5期
杨 斌 孟庆瑶 张 凯 段义忠
(榆林学院生命科学学院,榆林 719000)
叶绿体普遍存在于绿色植物和一些自养生物中,是植物细胞中的质体之一,主要进行光合作用,承担着合成蛋白质、色素、脂肪酸及淀粉等功能[1]。在被子植物中,叶绿体基因组一直处于相对保守的状态,导致其进化速度缓慢,被用于植物分类和分子进化研究的途径[2]。其结构大多为双链环形,具有4个区域,即1个大单拷贝区(Large single copy,LSC)、1个小单拷贝区(Small single copy,SSC)以及2个反向重复序列区(Inverted repeats,IRA/IRB)。由于生存环境和遗传差异的影响,不同种植物叶绿体基因组会出现结构变异的现象[3~5],常见的结构变异有缺失、重复、倒位和易位。
作为最常用的生物学分析方法之一,DNA测序为遗传信息的揭示和基因组分析等研究提供重要数据,对确定植物进化关系等方面发挥重要作用。“下一代”测序技术(Next-generation sequencing)是继第一代测序技术(Sanger)之后而得名的,主要包括3种测序技术:Roche/454焦磷酸测序(2005年)、Illumina/Solexa聚合酶合成测序(2006年)和ABI/SOLiD连接酶测序(2007年)技术,这3种测序技术与Sanger相比共同具有单次运行(Run)产出序列数据量大的特征,又被统称为高通量测序技术(High-throughput sequencing)。近年来,随着测序技术的发展,促使更多绿色植物的完整叶绿体全基因组相继公布在NCBI数据库,在2010年NCBI数据库中仅有146种,截止2019年4月已有3 000多种植物的叶绿体全基因组被记录[6]。
矮扁桃(Amygdalusnana)属蔷薇科(Rosaceae Juss.)植物,起源于欧洲东南部和亚洲中西部[7]。作为古地中海第三纪子遗物种,主要分布在哈萨克斯坦、俄罗斯和中国,不仅可作为油料和药物的原材料,还可以作为育种研究的原始材料。目前,国内外对于扁桃亚属(Subgen.Amygdalus)植物的研究主要集中在物质鉴定、生长发育、植物引种、开发利用和进化分析等方面[8~12]。在众多关于扁桃亚属研究中,基于叶绿体全基因组的相关研究比较缺乏。鉴于此,本研究对矮扁桃叶绿体全基因组进行测序、组装、注释和特征剖析,并与其他28种近缘物种构建系统发育树,旨在丰富矮扁桃的叶绿体基因组遗传信息,为今后桃属植物的系统进化关系研究和物种鉴定提供理论基础。
1 材料与方法
1.1 材料
矮扁桃新鲜叶片采集于新疆裕民县(46°05′51.02″N,82°48′9.06″E),处理后保存于-80℃环境中备用。
1.2 基因组的提取和测序分析
1.2.1 叶绿体DNA的提取
常见的高等植物叶绿体DNA提取的方法主要有3种:蔗糖密度梯度离心法、Percoll密度梯度离心法和高盐—低pH法,并已在高粱、甘蔗、苹果[13~15]等物种的叶绿体DNA提取中成功应用,未曾发现应用于矮扁桃的报道。由于叶绿体基因组的稳定性,在细胞质遗传、基因功能和遗传多样性等方面的研究广泛应用,而获取无污染、结构完整的叶绿体DNA则是开展相关研究的前提条件之一。因此本研究决定采用改良的蔗糖密度梯度离心法,便于提取完整的矮扁桃叶绿体DNA[16]。
1.2.2 叶绿体DNA测序分析
将提取的总叶绿体DNA通过Nano Drop 2000微量分光光度检测浓度是否符合标准,并用1%琼脂糖电泳检测质量。利用Invitrogen试剂盒对产物进行纯化,使所提取的叶绿体DNA达到高通量测序要求。最后将样品送至北京百迈客生物科技有限公司检测合格后,进行Illumina HiSeq Xten测序,得到的原始序列(Original series)数据以FASTQ格式存储。FASTQ文件中存在一定比例的低质量序列,滤除质量较低的序列末端、Adapter序列和含N比例达到10%的序列等[17],最终得到高质量的待分析序列(Clean Reads)。
1.3 基因组装与注释方法
对低质量过滤后的数据用SOAPdenovo 2.04拼接软件(http://soap.genomics.org.cn/soap denovo.html)组装成重叠群,再根据reads的paired-end和overlap优化组装结果。对于序列中的一些缺口(Gaps),通过Gapcloser对组装结果进行补洞和修正。最后利用BLAST(https://blast.ncbi.nlm.nih.gov/Blast.cgi)验证IR区和SC区的连接,进而获得完整的矮扁桃叶绿体全基因组。利用DOGMA在线注释软件(http://phylocluster.biosci.Ttexas.edu/dogma/)结合NCBI上已报道的蔷薇科近缘物种的注释结果对矮扁桃叶绿体全基因组进行基因注释,并通过GeneiousR8进行手动修正注释。最后利用OGDraw(https://chlorobox.mpimp-golm.mpg.de/OGDraw.html)[18]对矮扁桃叶绿体全基因组进行基因组图谱绘制。将新获得的矮扁桃叶绿体全基因组上传至GenBank数据库,登录号为MK764428。
1.4 重复序列与SSR分析
利用在线软件REPuter(https://bibiserv.cebitec.uni-bielefeld.de/reputer)[19],鉴定矮扁桃叶绿体全基因组中重复序列,包括正向重复(Forward repeats)、反向重复(Reverse repeats)、回文重复(Palindromic repeats)和互补重复(Complement repeats),最小重复长度设为20bp,两个重复之间的一致率大于90%。利用MISA软件(https://webblast.ipk-gatersleben.de/misa/index.php)鉴别矮扁桃叶绿体全基因组中简单重复序列(Simple sequence repeats,SSR),最小重复次数设为:单核苷酸≥10,二核苷酸≥6,三核苷酸≥5,四核苷酸、五核苷酸、六核苷酸都为≥3。
1.5 IR区边界的收缩和扩张分析
叶绿体全基因组的长度多态性主要由于IR区的扩张和收缩造成的,本研究比较矮扁桃、扁桃及其亲缘物种的IR边界区收缩与扩张情况,并利用IRscope(https://irscope.shinyapps.io/irapp/)绘制对比图[20]。
1.6 矮扁桃系统发育分析
通过NCBI数据库选取蔷薇科和虎耳草科(Saxifragaceae)共计28种植物的叶绿体全基因组与矮扁桃叶绿体全基因组进行系统发育分析。利用MAFFT软件[21]对29个序列进行多重序列比对,将比对结果检验和校正后导入MEGAX软件,以厚叶溲疏(Deutziacrassifolia)和黄脉绣球(Hydrangealuteovenosa)作为外类群,通过近邻结合法(Neighbor-joining,NJ)构建系统发育树,自举置信值基于重复抽样1 000次。
2 结果与分析
2.1 矮扁桃叶绿体全基因组的结构特征
矮扁桃的叶绿体全基因组长度为158 596 bp;呈现出经典的四段式环状结构,其中大单拷贝区(LSC)长度为86 771 bp,小单拷贝区(SSC)长度为19 037 bp,两个反向重复序列(IRA/IRB)长度都为26 394 bp。矮扁桃的叶绿体全基因组中CG含量为36.8%,在LSC区域中CG含量分别为34.5%,SSC区域中CG含量分别为30.3%,IR区域中CG含量分别为42.7%和42.6%(见表1)。矮扁桃的叶绿体全基因组共注释130个基因,其中蛋白编码基因(Protein-coding genes,PCGs)为85个,转运RNA(tRNA)为37个,核糖体RNA(rRNA)为8个(见表2)。在130个注释基因中,有92个为单拷贝基因,包括69个PCGs、23个tRNA基因;剩余19种基因均重复1次,包括PCGs中的ndhB、rpl2、rpl23、rps12、rps7、ycf15、ycf2、ycf1和tRNA基因中的trnA-UGC、trnI-CAU、trnI-GAU、trnL-CAA、trnN-GUU、trnR-ACG、trnV-GAC以及4种rRNA基因。其中LSC区域完全包含59个PCGs和22个tRNA基因,SSC区域包含12个PCGs和1个tRNA基因,而IR区域则包含7种PCGs(rpl2、rpl23、ndhB、rps7、ycf1、ycf15、ycf2)和7种tRNA基因(trnA-UGC、trnI-CAU、trnI-GAU、trnL-CAA、trnN-GUU、trnR-ACG、trnV-GAC)以及全部4种rRNA基因(见图1)。

表1 矮扁桃叶绿体全基因组碱基组成
2.2 叶绿体全基因组重复序列与SSR分析
微卫星DNA(Microsatellite DNA)又称简单重复序列(SSR),基本重复单位只有1~6 bp,存在于真核生物的细胞核、线粒体和叶绿体基因组中,在个体之间具有良好的通用性,被广泛地应用于物种鉴定及群体和个体之间的遗传差异分析。通过所设置参数,在矮扁桃叶绿体全基因组中共鉴定出71个SSR位点,其中单核苷酸重复数目为56,二核苷酸重复数目为3,无三核苷酸重复,四核苷酸重复数目为6,五核苷酸重复数目为2,没有六核苷酸重复,但有4个复合SSR位点(见表3)。在矮扁桃叶绿体全基因组的简单重复序列中有91.07%为AT类型,而CG类型仅有5个,且有66.20%的SSR位点位于IGS区,18.30%在内含子区,15.49%位于CDs中。
在矮扁桃的叶绿体全基因组中,检测出48条长度大于20 bp的重复序列,其中正向重复有16个,反向重复有8个,回文重复有23个(见表4)。仅有1个互补重复,位于trnT-UGU-trnL-UAA基因间隔区(IGS)。其中最长的重复序列为No.1,是回文重复序列,长53 bp,位于trnG-GCC-trnR-UCU基因间隔区;最短是正向重复序列No.47,仅长23 bp,位于ycf1基因上。

表2 矮扁桃叶绿体全基因组注释基因信息
注:(2)表示重复单元数为2
Note: (2)indicates that the number of the repeat unit is 2
2.3 IR区边界的收缩和扩张分析
矮扁桃叶绿体全基因组由4个经典区域组成,包括2个反向重复序列区(IRA/IRB)、1个大单拷贝区(LSC)和1个小单拷贝区(SSC)。在绿色植物长期发育与进化的过程中,IR区会出现与两边界的单拷贝区(LSC/SSC)重叠的现象,不同植物所表现的重叠长度的不同。我们选取矮扁桃的5种近缘种(扁桃Amygdaluscommunis;蒙古扁桃Amygdalusmongolica;桃Amygdaluspersica;大叶桂樱Laurocerasuszippeliana;沙梨Pyruspyrifolia)的叶绿体全基因组作为参考序列,比较分析矮扁桃的IR区边界的扩张和收缩情况(见图2)。分析结果显示6种蔷薇科近缘植物之间的叶绿体基因组结构差异较小,表现出叶绿体基因组高度的保守性。6种被子植物的叶绿体基因组结构相似,且都具有6个基因位于IR区边界处,分别是LSC/IRB的rpl22、rps19和rpl2,IRB/SSC的ndhF,SSC/IRA的ycf1,IRA/LSC的trnH。但仍存在一定的差异性,这6种植物的叶绿体基因组中rps19基因全部横跨LSC/IRB区,但在IRB区向rps19基因不同程

表3 矮扁桃叶绿体全基因组SSR预测
注: p.单个SSR类型;p1/p2/p3/p4/p5/p6中数字分别表示构成基序的碱基个数;c.复合SSR类型;*为位于内含子区
Note: p.Indicates single SSR type; The numbers in p1/p2/p3/p4/p5/p6 indicate the number of bases constituting the motif, respectively; c. Indicates composite SSR type;*is located in the intron region
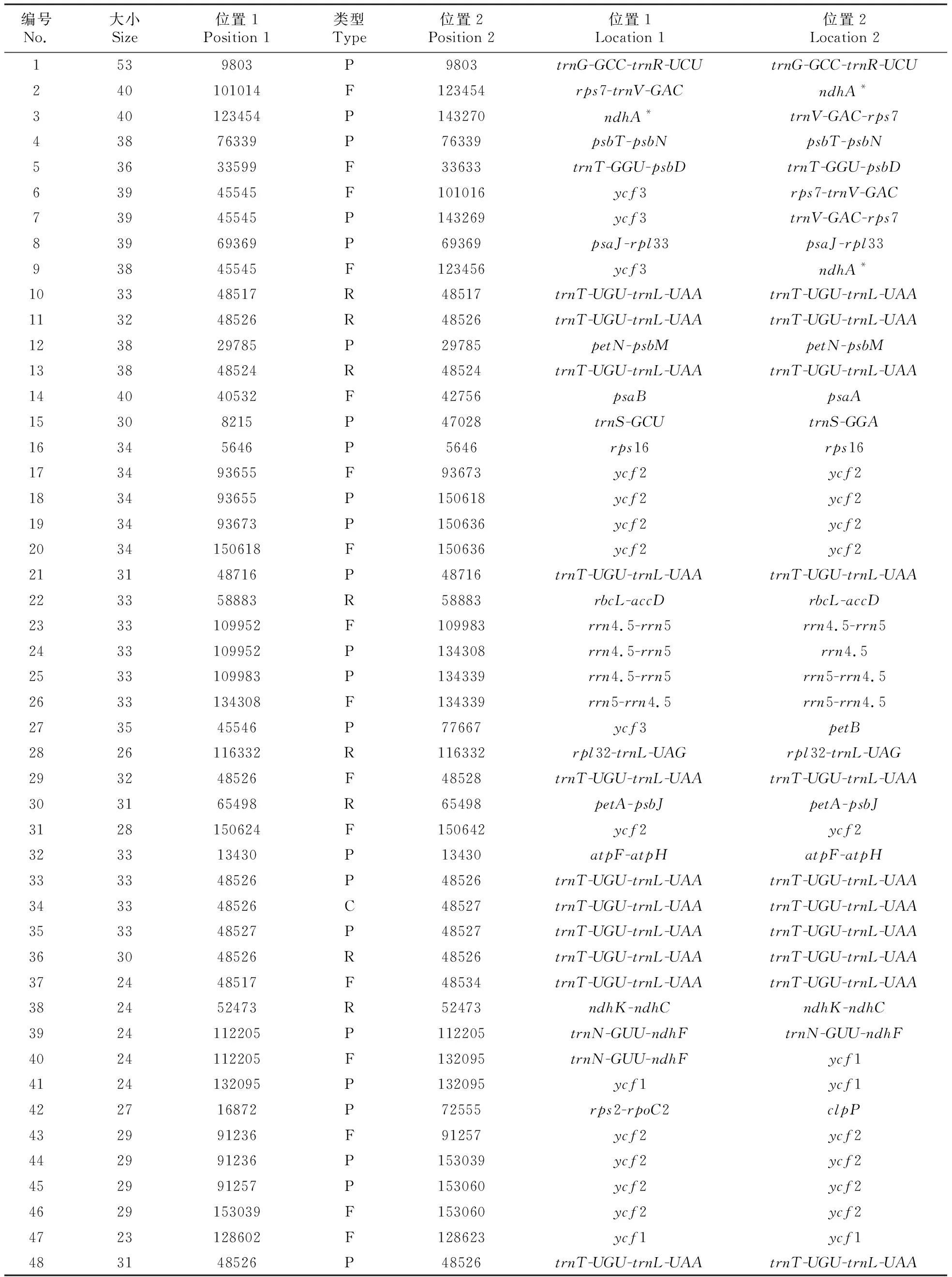
表4 矮扁桃叶绿体全基因组的重复序列
注:F.正向重复;R.反向重复;C.互补重复;P.回文重复;*.重复序列位于内含子区
Note: F.Direct repeat; R.Inverted repeat; C.Complementary repeat; P.Palindrome repeat;*is the repeat sequence located in the intron region.

图1 矮扁桃叶绿体全基因组图谱Fig.1 Chloroplast genome map of A.nana
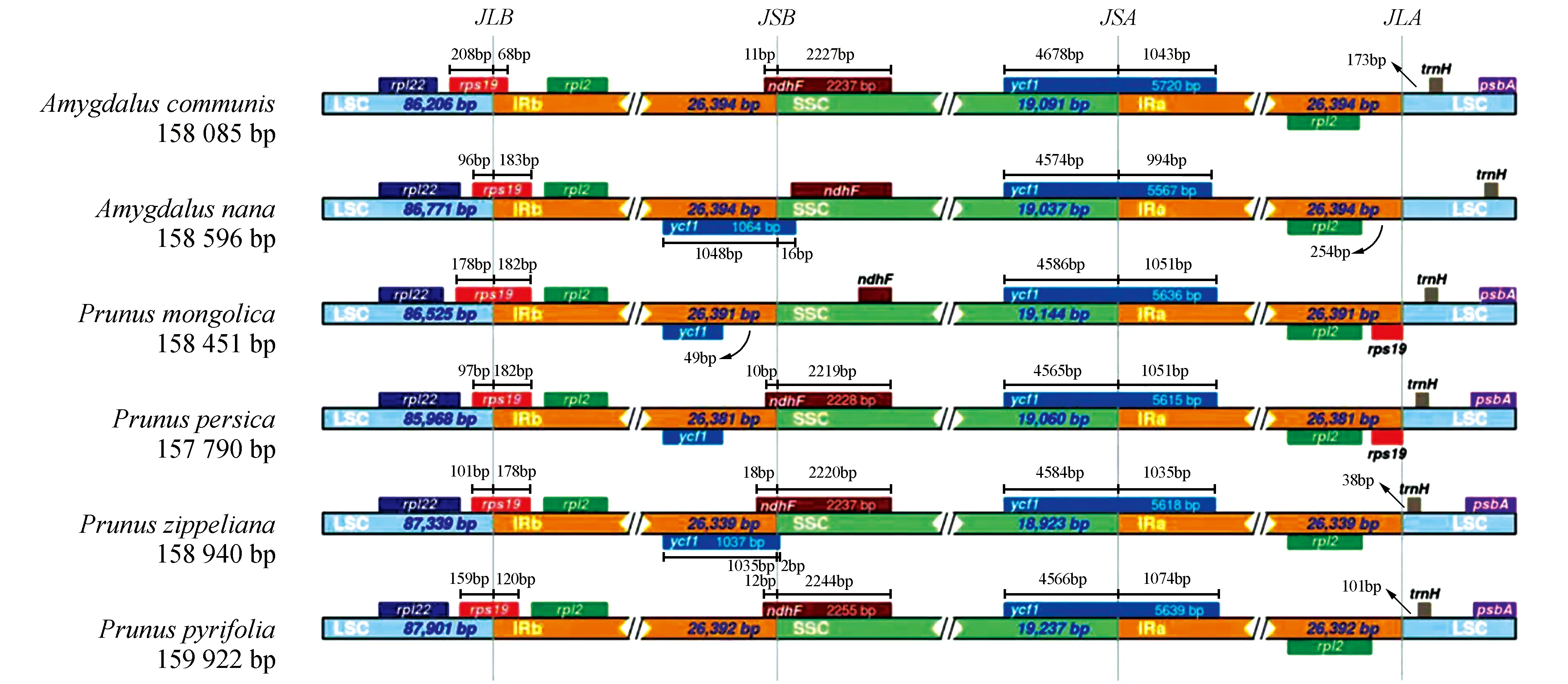
图2 6种植物的叶绿体IR区边界分析Fig.2 Boundary analysis of chloroplast IR region of 6 plants
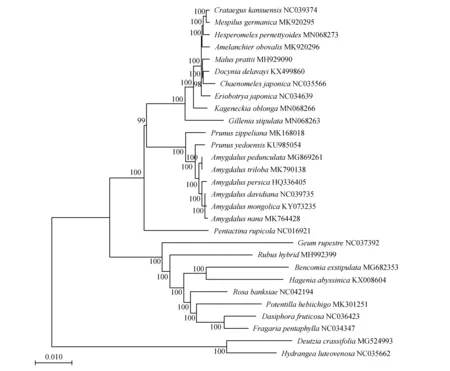
图3 基于29种植物构建的近邻结合树Fig.3 Nearest neighbor tree of 29 plants
度扩张了68~183 bp;在矮扁桃和蒙古扁桃叶绿体基因组中ndhF基因完全位于SSC区,而在其他的叶绿体基因组中IRB区发生了不同程度的扩张。在这6种植物的叶绿体基因组中trnH基因与IRA/LSC边界区的距离都有所不同,其中矮扁桃的距离最远。
2.4 叶绿体全基因组系统发育分析
选取矮扁桃在内的29种植物进行系统发育树的构建,以厚叶溲疏和黄脉绣球作为外类群,利用近邻结合法构建的发育树包含25个节点,低于100%的仅2个节点,其余节点均为100%,表明聚类结果可信度较高(见图3)。从系统发育树中发现矮扁桃、蒙古扁桃和山桃(Amygdalusdavidiana)以100%的支持率聚在同一分支上,而榆叶梅(Amygdalustriloba)和长柄扁桃(Amygdaluspedunculata)所构成的单系支持率也为100%。在扁桃亚属中,矮扁桃在亲缘关系上与蒙古扁桃更近,而与长柄扁桃和榆叶梅的亲缘关系稍远。
3 讨论
被子植物的叶绿体基因组具有典型结构,一般为闭合环状的双链DNA并包括4个高度保守的区域,其大小通常为115~165 kB,编码为110~130个基因[22]。4个结构区域长度范围通常为:IR区(16~27 kB)、LSC区(80~90 kB)和SSC区(20~28 kB)。尽管大多是植物的叶绿体基因组结构保守,但是在各区域大小上却不尽相同,如在木犀科(Oleaceae)植物中测得SSC区长度在13 252~17 908 bp[23]。本研究矮扁桃的叶绿体全基因组序列长度为158 596 bp,包括IRs区(26 394/26 394 bp)、LSC区(86 771 bp)和SSC区(19 037 bp),为进一步确定被子植物叶绿体基因组的各结构区域长度范围提供参考依据。同时对包括矮扁桃在内的6种植物进行IR边界区分析,发现6种植物的rps19基因全部横跨LSC/IRB区,这与5种已知的菊科(Compositae)植物的IR区分析结果相同[24],不仅降低了本研究结果的偶然性,也体现出被子植物叶绿体基因组的保守性。叶绿体基因组的SSR标记不仅保留该基因组SSR标记的特点,而且有较高的重复性,可作为种质鉴定的一种途径。本研究中的矮扁桃的叶绿体全基因组中共有71个SSR位点,其数量要远大于禾本科(Gramineae)植物[25],而小于桑科(Moraceae)植物[26];并发现其中未涉及三核苷酸重复类型,然而在其他被子植物的叶绿体全基因组中却存在三核苷酸重复类型[27]。
为进一步确定矮扁桃在被子植物中的进化地位和亲缘关系,选取NCBI已公布的28种植物,以厚叶溲疏和黄脉绣球作为外类群进行系统进化分析。本研究中榆叶梅和长柄扁桃以100%的支持率聚于同一分支,这与邱蓉和程中平等人通过传统植物学性状和核质DNA分析长柄扁桃和榆叶梅亲缘关系的研究结果一致[28]。除此之外,矮扁桃、蒙古扁桃和山桃聚在同一分支上,并且矮扁桃与同属的蒙古扁桃关系最近,这与邱蓉基于ITS序列构建的系统发育树相吻合[29]。
利用矮扁桃在内29种植物叶绿体全基因组序列数据构建系统发育树,为桃属(Amygdalus)植物之间的进化途径及近缘物种间的亲缘关系提供有力的证据,同时对矮扁桃叶绿体基因组的分析为其在分子标记开发和分子鉴定等研究提供参靠数据。
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22 00:33:26
肝博士(2022年3期)2022-06-30 02:48:28
Journal of Sport and Health Science(2019年6期)2019-11-26 07:30:53
中成药(2018年6期)2018-07-11 03:01:20
甘肃林业(2016年3期)2016-11-07 08:56:28
国外医药(抗生素分册)(2016年2期)2016-07-12 14:25:01
广西林业科学(2016年1期)2016-03-20 05:33:01
癌变·畸变·突变(2014年2期)2014-03-01 04:39:42
食品科学(2013年6期)2013-03-11 18:20:13
中国烟草学报(2012年2期)2012-04-09 06:44:56
