
婴儿尼曼-匹克病1例
2020-06-29 09:15周光德
临床与实验病理学杂志 2020年5期
龙 梅,王 琪,周光德
患儿女婴,8个月,因腹部膨隆8个月,咳嗽、流涕5天入院。患儿系第3胎第3产,母亲孕期无异常,37+3周剖腹产(瘢痕子宫),生后无窒息史,生长发育如同龄儿童,智力正常。家簇史:父母非近亲结婚,其同胞兄长因肝脾肿大原因夭折。入院后体检:生命体征平稳,双肺呼吸音粗,可闻及中细湿性啰音。心脏无异常,全腹无压痛、反跳痛,肝脏于肋下7.5 cm、剑下6 cm扪及,质韧、边钝,脾脏肿大(甲乙线8 cm,甲丙线9 cm,丁戊线2 cm),质韧、边钝,移动性浊音阴性,肠鸣音4次/分,神经系统检查未引出病理性神经反射。实验室检查:肝功能ALT 714 U/L,AST 642 U/L,ALP 368 U/L,TBIL 17.31 μmol/L,DBIL 6.75 μmol/L,ALB 44.9 g/L;腹部B超(7.18):肝大、脾大;胸部CT示双肺透亮度降低,充气不均匀,双肺纹理增多、增粗、紊乱、模糊;其间多发结节影及斑片影,边缘模糊(图1);基因检测:基因编码c.1458T>G(胸腺嘧啶>鸟嘌呤),c.1498T>C(胸腺嘧啶>胞嘧啶),2处杂合突变(图2)。
病理检查眼观:肝脏穿刺组织一块,淡黄色,大小1.0 cm×0.1 cm×0.1 cm。镜检:肝细胞弥漫性肿胀,胞质泡沫状,糖原染色阳性(图3);骨髓镜检:见泡沫细胞,直径20~100 μm,核圆形或椭圆型,单核或双核,胞质丰富,充满桑椹状或泡沫状透明小泡,糖原染色阳性(图4)。
病理诊断:尼曼-匹克病(Niemaoh-Pick disease, NPD)。
讨论NPD又称鞘磷脂沉积病(sphingomyelin lipidosis),属先天性糖脂代谢性疾病,是一组脂质代谢异常的溶酶体贮积病[1]。其特点是全单核巨噬细胞和神经系统有大量的含有神经鞘磷脂的泡沫细胞。为常染色体隐性遗传[2],以犹太人发病较多,其发病率高达1/25 000。
NPD临床上常以肝、脾肿大和神经系统受损,肺部感染为主要表现,因临床表现多样,将本病分为A-E3型[3]、NPD-A型。NPD-A型即婴儿型,是最常见的类型,出生后数周出现肌力及肌张力低下,神经系统症状出现较早,病情进展迅速,多数患儿3岁左右死亡。NPD-B型,即慢性非神经型,发病较A型晚,临床特征仅涉及内脏器官,脾脏先增大,后出现肝脏增大,病情进展缓慢,因肺部弥漫性浸润而容易发生感染,一般不影响寿命。NPD-C型即慢性神经型,多数在儿童期或青春期发病,病初表现为运动发育迟缓,构音障碍,继而出现共济失调、意向性震颤等[4]。该患儿骨髓细胞学检查及肝脏病理学检查均找到泡沫细胞,即尼曼-匹克细胞,结合该患儿幼年发病,出生后出现腹胀,进行性加重,以脏器浸润为主,表现为肝大、脾大,肝功能异常,肺部感染为主要表现,胸部CT示粟粒状浸润,临床检查除外结核杆菌及支原体感染,故临床诊断为NPD。
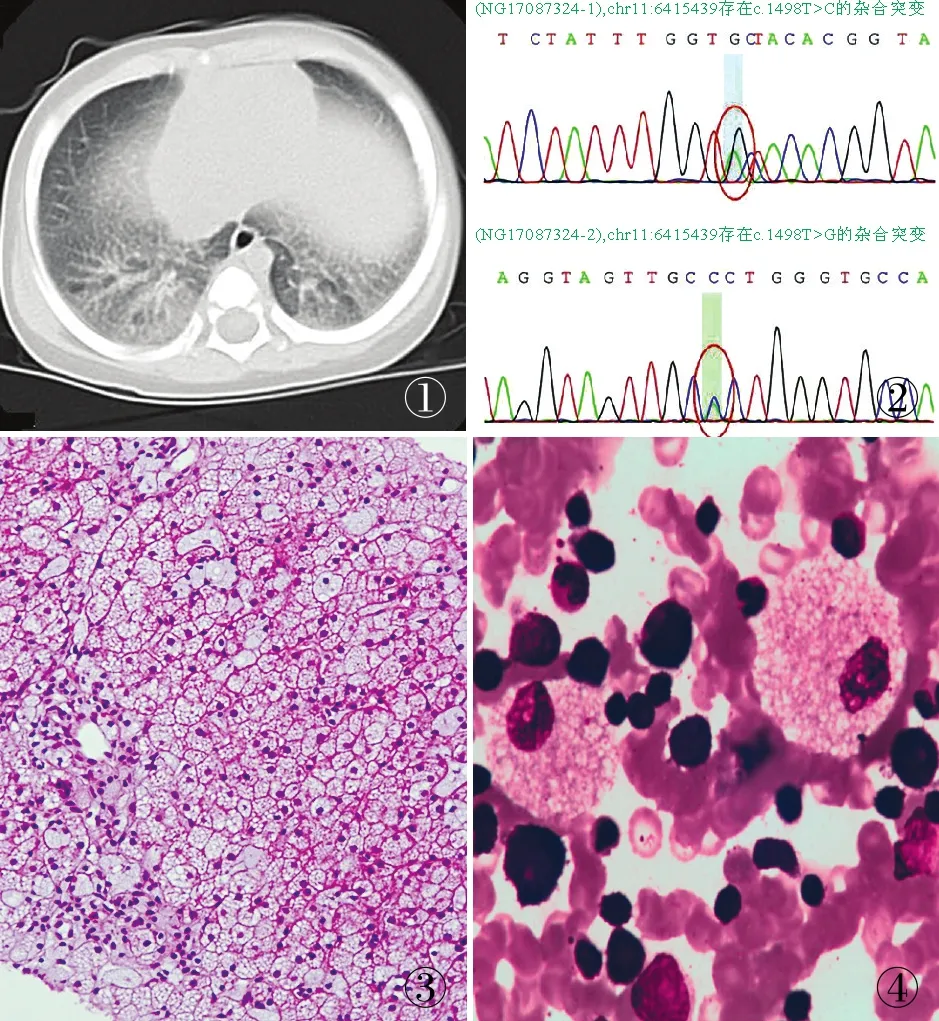
图1 肺部CT示双肺透亮度降低,充气不均匀,双肺纹理增多、增粗、紊乱、模糊;其间多发结节影及斑片影,边缘模糊 图2 基因编码:c.1458T>G(胸腺嘧啶>鸟嘌呤),c.1498T>C(胸腺嘧啶>胞嘧啶),2处杂合突变 图3 肝细胞弥漫性肿胀,胞质泡沫状,糖原染色阳性 图4 骨髓镜检见泡沫细胞,直径20~100 μm,核圆形或椭圆型,单核或双核,胞质丰富,充满桑椹状或泡沫状透明小泡,糖原染色阳性
目前已知A型和B型均是由于SMPD1基因突变导致其编码的酸性鞘磷脂酶(ASM)活性下降或缺失,引起溶酶体内酸性鞘磷脂沉积,所以又合称为酸性鞘磷脂酶缺陷病,NPD-C型(NPC)是由于NPC1或NPC2基因突变所致,主要病理改变是组织细胞内大量游离胆固醇沉积[5],血神经鞘磷脂酶活性测定、尿神经鞘磷脂排泄量测定、骨髓检查、肝脾或淋巴结活检及基因分析[6],该患儿基因检测提示NPD-A型、NPD-B型相关基因SMPD1存在2处杂合突变,结合该患儿神经系统不受累及,无肌力及肌张力低下,无生长发育及智力发育落后,无反复腹泻、呕吐及喂养困难,故目前考虑该患儿为NPD-B型。
由于NPD发病率低,易漏诊或误诊,组织学检查发现大量泡沫样细胞,可提高诊断的准确性,但需结合基因及临床资料综合判断,早期明确诊断及临床分型,改善预后。目前该病尚无有效的治疗方法,主要是对症治疗为主,改善患者的脏器及神经功能,提高生存质量。
猜你喜欢
广西糖业(2022年5期)2022-11-24
云南医药(2019年3期)2019-07-25
实用肿瘤学杂志(2019年5期)2019-02-10
成都体育学院学报(2017年1期)2017-02-21
天津医药(2016年9期)2016-10-20
现代工业经济和信息化(2016年3期)2016-05-17
中国老年学杂志(2015年16期)2015-03-05
癌变·畸变·突变(2015年4期)2015-02-27
植物营养与肥料学报(2011年4期)2011-10-26
求医问药(2009年7期)2009-08-31
