
制备方法对FeC2O4制备的费托合成铁催化剂的影响
2020-06-29 04:12罗明生冯旭楞宋丹杨智王亚涛李洪娟
化工进展 2020年6期
罗明生,冯旭楞,宋丹,杨智,王亚涛,李洪娟
(1 北京石油化工学院化学工程学院,燃料清洁化及高效催化减排技术北京市重点实验室,北京102617;2 开滦煤化工研发中心,河北唐山063611)
费托合成(FTS)是将煤、天然气以及生物质等含碳资源生产的合成气(H2+CO)转化为液态燃料和高附加值产品的过程。传统的费托合成工业铁基催化剂常以硝酸铁[Fe(NO3)3]为铁前体进行制备,然而在制备的过程中会产生大量的NO-3,造成环境污染,因此寻求环保可替代的铁前体对于费托合成铁催化剂的发展至关重要。一些学者也在铁前体对催化剂结构和性能的影响方面进行了许多研究和探索[1-9],研究发现不同铁前体制备的催化剂具有不同的催化性能。
本文引入了一种新的铁前体草酸亚铁(FeC2O4),通过并流共沉淀、正加共沉淀、沉淀-浸渍K和沉淀-浸渍K、SiO2四种方法来制备100Fe/4Cu/4K/16SiO2铁催化剂,使用扫描电子显微镜(SEM)、比表面积测量仪(BET)、X射线衍射仪(XRD)、程序升温还原(TPR)等表征手段,对催化剂的表面化学性质和体相结构进行了讨论分析,并结合性能评价结果,对比出最佳的制备方法,以期对费托合成铁催化剂的发展进行新的探索。
1 实验部分
1.1 催化剂制备
以并流共沉淀方法为例,取20g FeC2O4·2H2O(阿拉丁,AR)和0.94g Cu(NO3)2·3H2O(国药集团,AR)配成400mL混合溶液(质量比Fe∶Cu=100∶4),并将两者搅拌均匀,放置于75℃的水浴中预热。配制1mol/L 的Na2CO3溶液,放置于75℃的水浴中预热。反应开始时,将混合溶液与沉淀剂同时并流滴加到有20mL 去离子水的大烧杯中,将水浴温度控制在80℃,滴加和搅拌同时进行,并控制pH 稳定在8.57(±0.02),直至混合溶液反应完全。沉淀完成后在80℃水浴中继续搅拌1h,之后静置老化3h。过滤洗涤后,将滤饼重新打成浆液,将0.4406g K2CO3(天津福晨化学试剂厂,AR)、3.3205g 硅溶胶(广州穗欣化工有限公司,工业级,含量30%)加入到浆液中,剧烈搅拌6h,过滤后将滤饼在100℃干燥12h,500℃下焙烧6h,将制得的催化剂进行压片筛分,即可得到质量比为100Fe/4Cu/4K/16SiO2的铁催化剂。正加共沉淀是将沉淀剂加入到金属盐溶液中;沉淀-浸渍K和沉淀-浸渍K、SiO2两种方法则是后期将K助剂、硅溶胶以浸渍法添加到并流共沉淀制备的铁前体中。制备过程如图1所示。
1.2 催化剂表征

图1 制备过程示意图
催化剂的织构性质由日本BELSORP-max 型全自动多站比表面、微孔和介孔孔隙分析仪测定。样品比表面积采用Brunauer-Emmet-Teller(BET)方法计算得出,样品的孔结构根据BJH模型测得。
催化剂的氧化还原性由日本BELCAT-B 型全自动程序升温化学吸附仪测定,将约50mg 催化剂置于石英反应管中,用H2∶Ar=1∶9混合气(30sccm)进行程序升温还原,以10℃/min 的升温速率升至800℃,保持30min,TCD检测H2的消耗量。
样品的物相结构采用日本岛津XRD-7000 型X射线衍射仪测定,Cu 靶Kα(λ=0.154nm),管电流30mA,管电压40kV,扫描范围10°~80°,扫描速度4°/min,扫描步长0.02°。
采用美国QUANTE400F 型扫描电子显微镜观测催化剂的表面微观/亚微观形貌。催化剂的形貌颗粒尺寸用Nano measurer 1.2软件进行分析。
1.3 催化剂评价
采用固定床反应装置对费托合成铁催化剂进行催化性能评价。反应时取60~80 目的催化剂颗粒1.0g填充于不锈钢反应管的中层恒温段,并加入一定量的SiC 颗粒进行散热作用。催化剂首先在纯CO 气氛下进行还原处理24h,其还原条件为:温度270℃、压力0.1MPa、空速3000mL/(gcat·h)。还原完成后,在270℃、1.5MPa、H2/CO体积比为1、空速3000mL/(gcat·h)的条件下连续反应120h。反应中气相产物通入Micro GC490 安捷伦气相色谱分析仪进行实时反应检测分析,液相产物和固相产物通过Varian SCION 436-GC离线气相色谱进行产物分析。催化剂的活性和产物的选择性通过外标法进行计算。
催化剂活性和产物选择性计算如式(1)~式(4)。CO转化率公式

CO2选择性公式

低碳烃类产物选择性

重质烃产物选择性
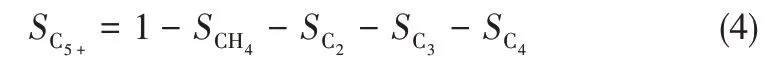
式中,XCO为CO的转化率;C[CO]in为进入反应器前CO 的初始浓度;C[CO]out为反应尾气中CO 的浓度;C[CO2]out为出反应尾气中CO2的浓度;SCx(x=1,2,3,4)为C1~C4低碳烃类的产物选择性;SC5+为重质烃产物的选择性。
2 结果与讨论
2.1 制备方法对外观形貌的影响
图2为不同方法制备的铁催化剂的SEM图,图3为催化剂的粒径分布情况。从图2可以看出,制备方法对铁催化剂的外观形貌有明显影响。从图2(a)可以看出,由并流共沉淀制备的催化剂为小的长条棒状结构堆砌在一起的块状结构,且小的棒状结构之间缝隙明显,小棒状颗粒平均宽度35.08nm,平均长度146.15nm。由沉淀-浸渍K、SiO2制备的催化剂形貌与并流共沉淀制备的催化剂形貌相似,颗粒的缝隙均不规整,颗粒的平均宽度48.45nm,平均长度174.76nm,较并流共沉淀制备的催化剂颗粒略大,说明浸渍的K、SiO2主要包覆在催化剂表面,导致颗粒的长大。由正加共沉淀制备的催化剂为28~132nm 的无规则块状颗粒,平均颗粒大小57.38nm,且块状表面布满了较小晶粒,推测催化剂的结晶不完善,结晶度较低。从图2(c)可以看出,由沉淀-浸渍K 制备的催化剂为很薄的层片状结构,直径为51~690nm,平均厚度16nm,可明显看出由沉淀-浸渍K 方法制备的催化剂有较高的比表面积。通过以上研究对比,说明制备方法对铁催化剂的结晶过程有重要影响。

图3 催化剂的粒径分布情况
2.2 制备方法对织构性质的影响
图4 为不同方法制备铁催化剂的吸附脱附曲线,可以看出不同方法制备的铁催化剂的吸附-脱附等温曲线均为Ⅳ型吸附等温线[10],吸附回滞环出现在p/p0≥0.7,这是因为催化剂内出现毛细凝聚现象,说明4种催化剂均为介孔材料。等温曲线中出现的吸附回滞环均为H3型[11],一般H3型回滞环等温线没有明显的饱和吸附平台,表明孔结构很不规整。H3 型反映的孔包括平板狭缝结构、裂缝和楔形结构等,这与SEM测试的结果一致。

图4 不同方法制备铁催化剂的吸附-脱附曲线
表1为不同方法制备铁催化剂的织构性质,可以看出4 种催化剂的平均孔径介于2~50nm 之间,属于介孔材料。根据比表面积的大小排序为沉淀-浸渍K>并流功沉淀>正加共沉淀>沉淀-浸渍K、SiO2,经过分析可以发现,比表面积的大小与催化剂的形貌有直接关系。

表1 不同方法制备铁催化剂的织构性质
图5为不同方法制备铁催化剂的孔径分布曲线图,可以看出由正加共沉淀、并流共沉淀和沉淀-浸渍K 三种方法制备的催化剂的孔径分布曲线相似,且孔径峰值均在6.12nm 处,说明由这三种方法制备的催化剂的平均孔径相近。而由沉淀-浸渍K、SiO2方法制备的催化剂的孔径峰值出现在14.03nm处,峰宽较大,相较于其他三种方法,该方法制备的催化剂平均孔径较大,可能是因为该方法分别两次浸渍的K和SiO2使催化剂的粒径明显增大,引起了孔径的变化。催化剂的孔径分布情况与BET测试的结果相一致。
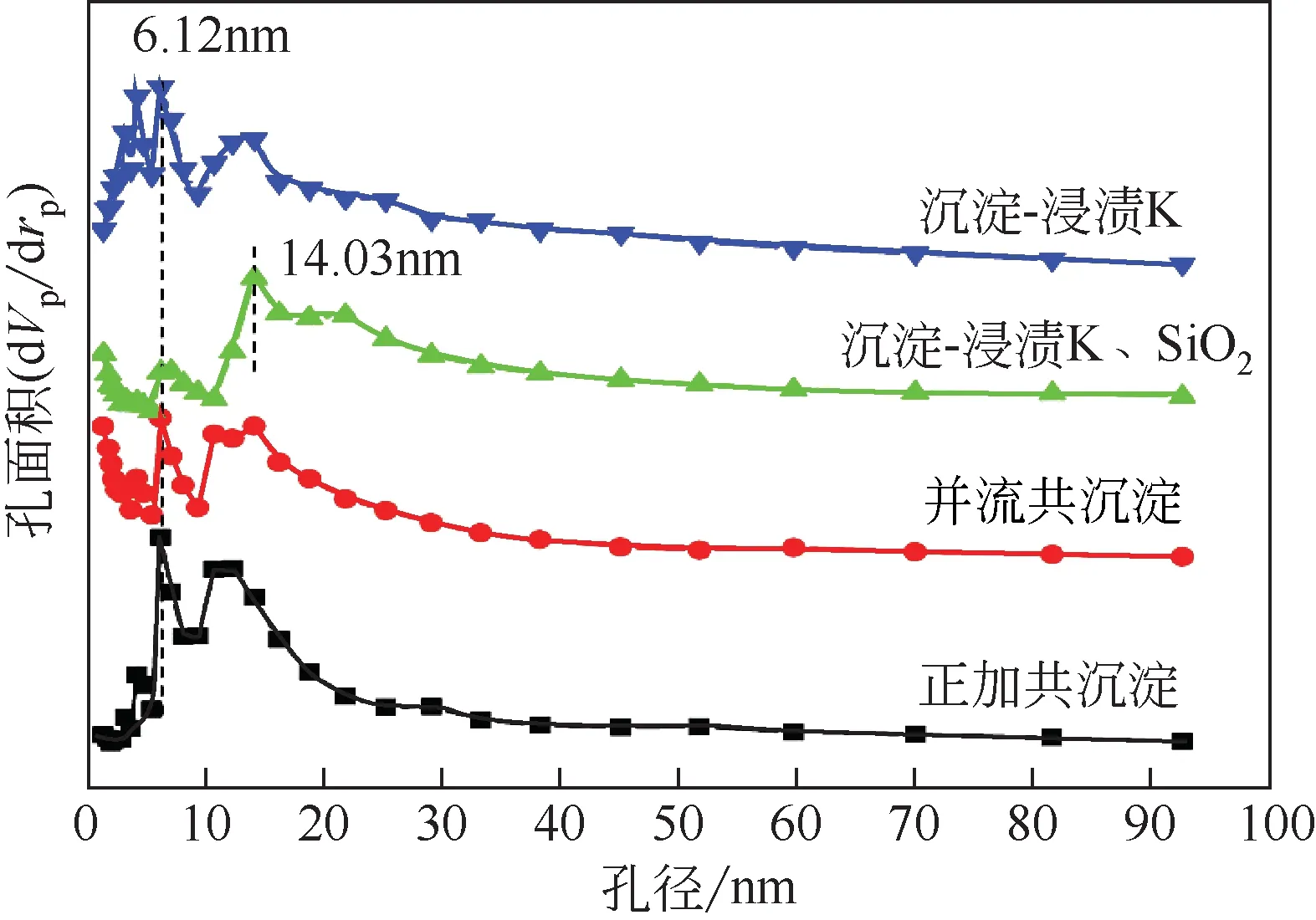
图5 不同方法制备铁催化剂的孔径分布曲线
2.3 制备方法对物相结构的影响
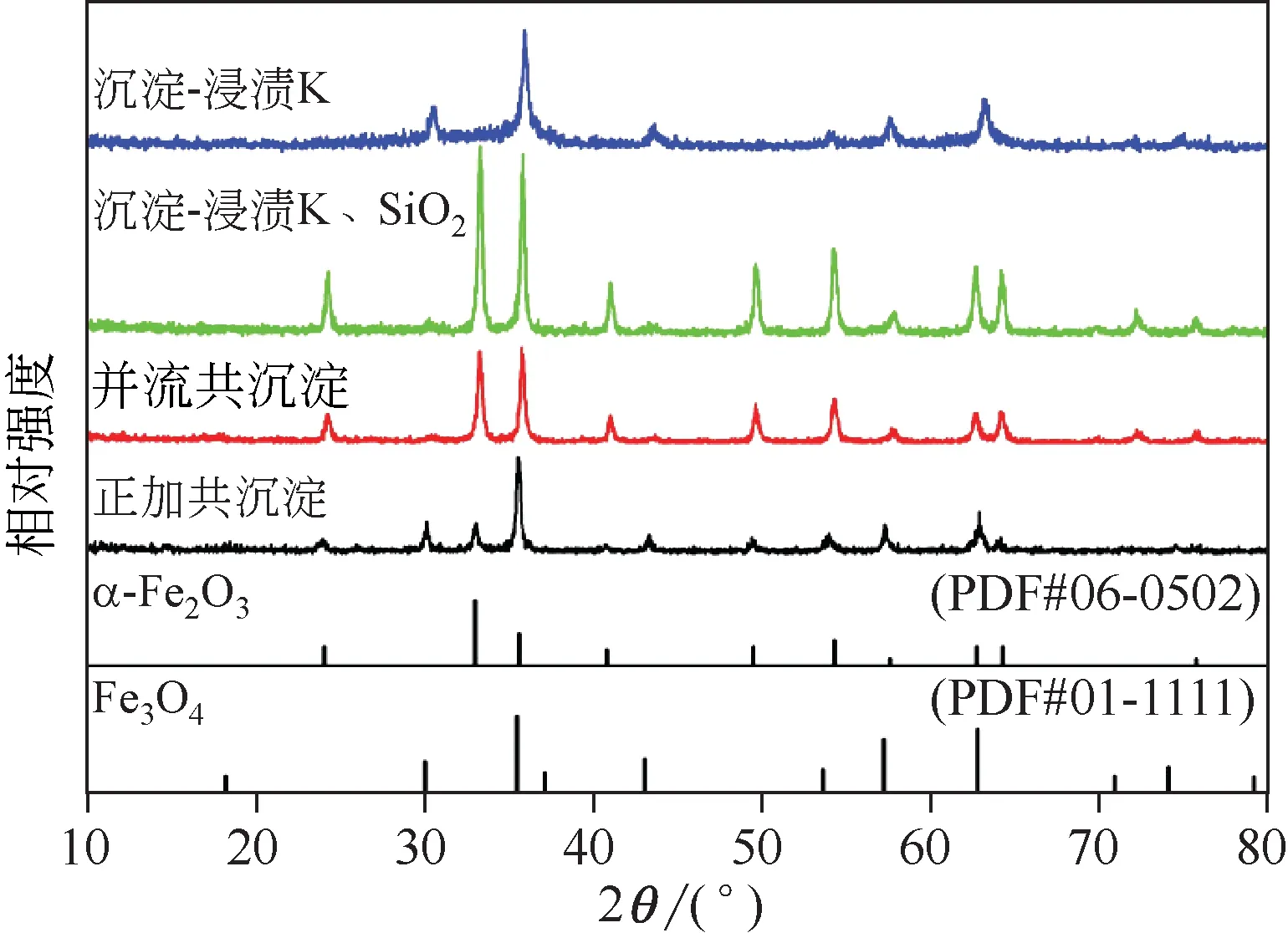
图6 不同方法制备铁催化剂的XRD图
图6为不同方法制备铁催化剂的XRD图,从图中可以看出4 种方法制备的催化剂均包含α-Fe2O3和Fe3O4的特征峰,说明4 种催化剂的物相为α-Fe2O3和Fe3O4的混合物。制备催化剂时,铁源中Fe2+会发生氧化变成Fe3+,使催化剂在焙烧过程中形成Fe2O3,未被氧化的Fe2+则形成了Fe3O4[4]。而图中α-Fe2O3和Fe3O4的衍射峰在强度和宽度上有明显的差异,说明催化剂中Fe 相的组成和晶粒大小受制备方法的影响。
催化剂中Fe2O3和Fe3O4的晶粒尺寸由谢乐公式计算得出,结晶度由Jade6 软件进行计算。不同方法制备的铁催化剂晶粒尺寸及结晶度如表2 所示。可见,催化剂的晶粒大小顺序与SEM 中颗粒大小顺序基本一致。制备方法对催化剂结晶度大小顺序为并流功沉淀>沉淀-浸渍K>沉淀-浸渍K、SiO2>正加共沉淀。对比发现正加共沉淀方法在制备铁催化剂前体的过程中的不同,会导致催化剂的均匀性较差,结晶不完善,从而影响催化剂的结晶度,与SEM分析结果一致。

表2 不同方法制备的铁催化剂的晶粒尺寸及结晶度
2.4 制备方法对还原性能的影响
图7 为不同方法制备铁催化剂的H2-TPR 图,可以看出4种催化剂在H2中均有低温和高温两个明显的还原阶段。在低温还原阶段(150~450℃),主要是催化剂中的Fe2O3或CuO 还原为Fe3O4或Cu;在高温还原阶段(600~800℃),Fe3O4还原为FeO或Fe[12-13]。一般来说,低温还原阶段包括三个小峰:第一个小峰是CuO 的还原峰(150~200℃),第二个小峰是CuO 和一部分Fe2O3紧密结合后的还原峰(200~320℃),第三个小峰则是单独的Fe2O3的还原峰(350~420℃)[14]。
从图7中可以看出,制备方法对铁催化剂在H2下的还原有显著影响。根据Fe2O3峰在H2下的还原温度,可以知道铁催化剂在低温下易还原程度顺序为并流共沉淀>沉淀-浸渍K>正加共沉淀>沉淀-浸渍K、SiO2。这主要是由于制备方法影响了助剂与Fe 相的结合方式。由并流共沉淀制备的催化剂中Cu 助剂明显促进了Fe2O3的还原,使Fe2O3的还原峰温度向低温方向发生偏移。而由沉淀-浸渍K、SiO2方法制备的催化剂则由于SiO2浸渍在了Fe相的外表面,增大了颗粒直径,抑制了Fe2O3的还原,使Fe2O3的还原峰温度最高。由沉淀-浸渍K方法制备的催化剂与正加共沉淀相比,Fe相表面浸渍的K助剂有利于催化剂的还原[15-16],所以由沉淀-浸渍K方法制备的催化剂的Fe2O3还原峰温度相对较低。

图7 不同方法制备铁催化剂的H2-TPR图
至于Fe3O4在高温阶段的还原,从图7 中可以看出,制备方法对其影响较小,整个还原过程均在600~800℃完成,说明制备方法对Fe3O4的还原几乎没有影响。
2.5 制备方法对催化性能的影响
表3 是不同方法制备的铁催化剂在反应60h 和120h 时的反应活性和产物选择性,通过对比催化剂的CO转化率,发现催化剂反应活性大小顺序为:并流共沉淀>沉淀-浸渍K>沉淀-浸渍K、SiO2>正加共沉淀。一般认为催化剂的活性与催化剂的分散性和还原性有直接关系。根据H2-TPR的表征结果,催化剂的还原性能顺序为:并流共沉淀>沉淀-浸渍K>正加共沉淀>沉淀-浸渍K、SiO2。根据BET的测试结果,比表面积的大小顺序为:沉淀-浸渍K>并流功沉淀>正加共沉淀>沉淀-浸渍K、SiO2。可见并流共沉淀和沉淀-浸渍K 两种方法制备的催化剂由于具有较好的低温还原性能及较高的比表面积,展现出较高的催化活性,在运行120h后CO 转化率分别为96.2%、91.8%。从图8 可以看出,正加共沉淀制备的催化剂初期(前35h)活性高于沉淀-浸渍K、SiO2方法。35h后,正加共沉淀制备的催化剂失活速率较快,稳定性远不如沉淀-浸渍K、SiO2方法。120h 后,正加共沉淀和沉淀-浸渍K、SiO2方法制备的催化剂CO 转化率分别为71.9%、83.8%。原因可能是正加共沉淀制备的铁催化剂的比表面积和还原性均略高于沉淀-浸渍K、SiO2方法,使正加共沉淀制备的催化剂初期活性较高,后期由于正加共沉淀制备的催化剂均匀性较差、结晶度低、活性相易失活等原因导致催化剂稳定下降。沉淀-浸渍K、SiO2方法制备的催化剂的活性相由于K和SiO2浸渍包覆,同时抑制了催化剂的还原和活性相的氧化,导致该催化剂前期催化活性低,后期稳定性强。
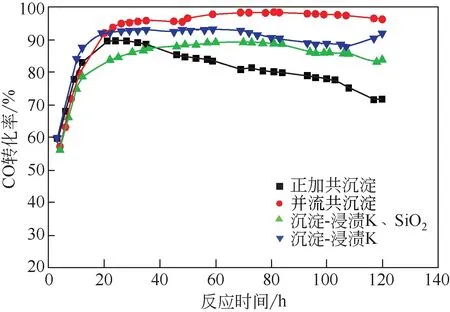
图8 CO转化率图(270℃,1.5MPa,3000mL/(gcat·h),H2/CO=1∶1)

表3 不同方法制备的催化剂的反应活性和产物选择性

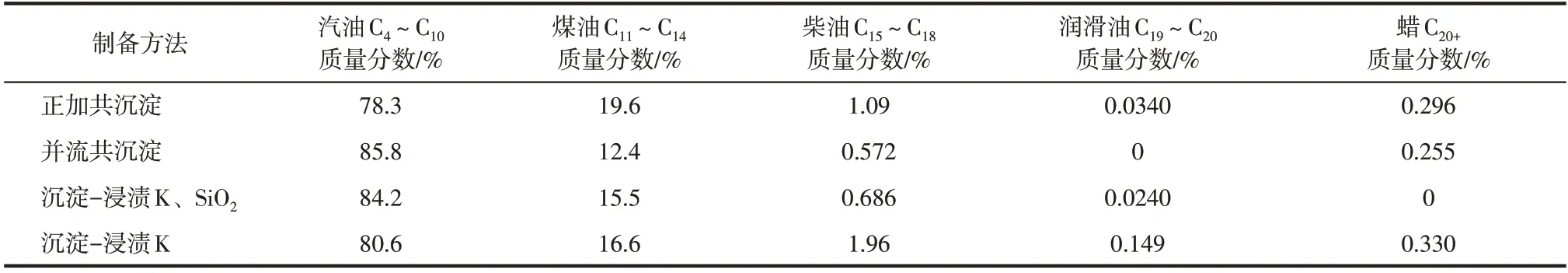
表4 制备方法对油相产物碳数分布的影响
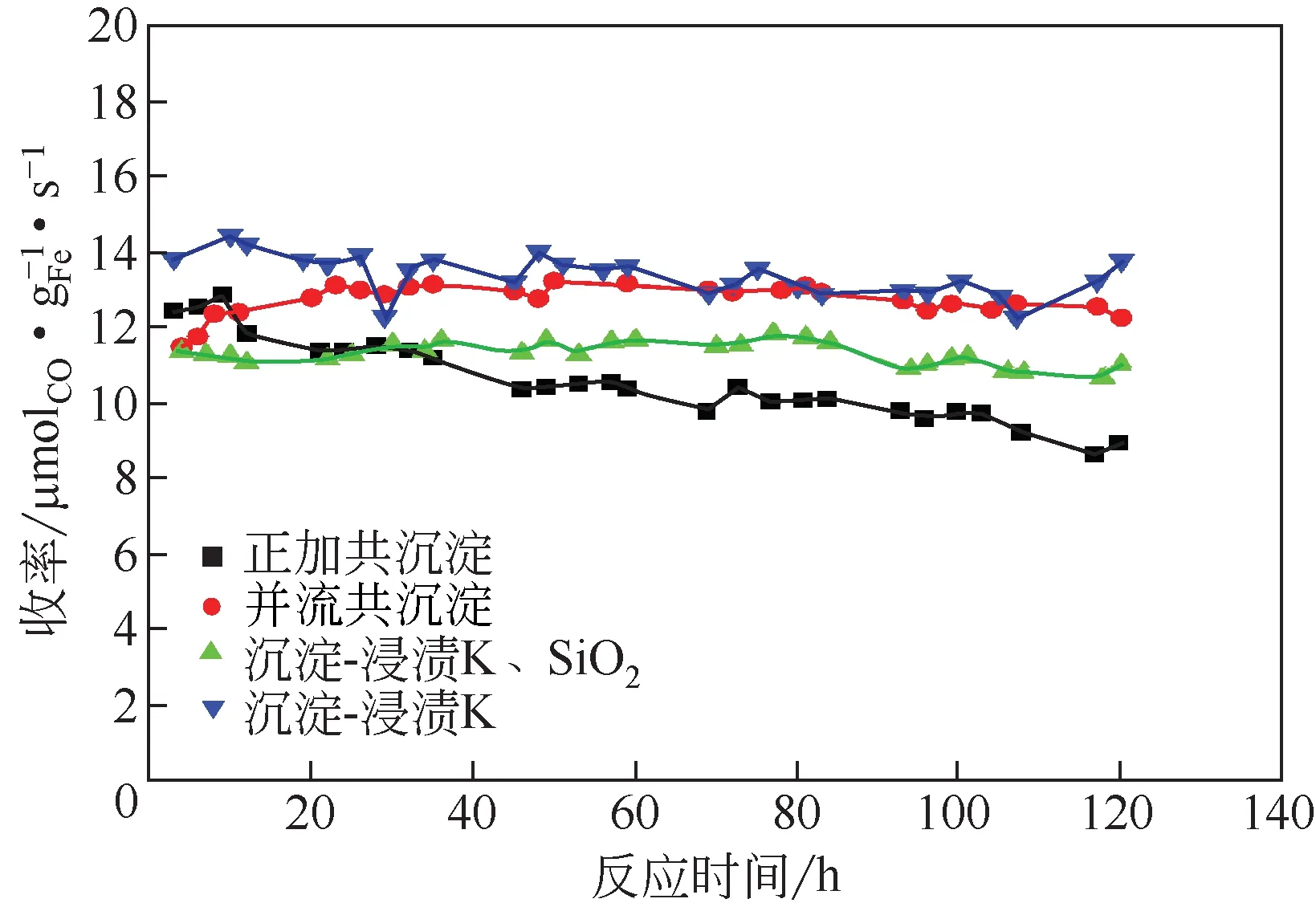
图9 产物收率图
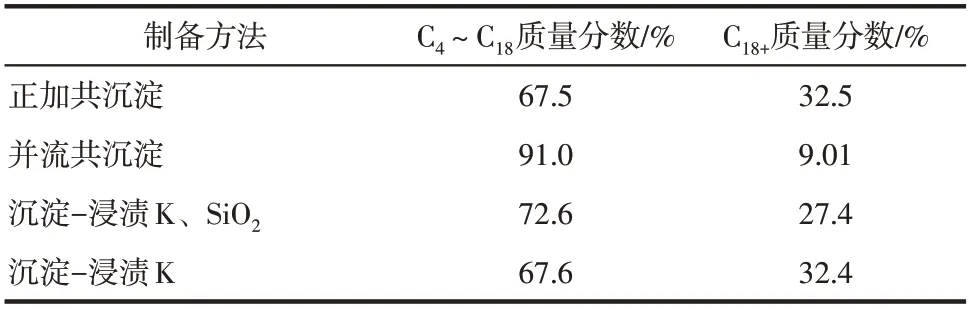
表5 制备方法对重质烃碳数分布的影响
从图8 可知,4 种催化剂的稳定性顺序为:并流共沉淀>沉淀-浸渍K>沉淀-浸渍K、SiO2>正加共沉淀。反应过程中的活性组分的氧化、积炭、烧结、流失、中毒以及被非活性组分掩盖等均会使催化剂失活[1]。本次研究中,催化剂的失活可归因于制备方法使助剂与Fe 相之间结合方式不同,导致活性位的氧化和掩盖。与并流共沉淀相比,正加共沉淀在制备铁前体的过程中的方式不同,导致Fe 相和Cu 相先后沉淀析出,甚至会出现反应不充分的情况,导致组分流失,使催化剂易氧化失活。而沉淀-浸渍K和沉淀-浸渍K、SiO2两种方法则由于K和SiO2的添加方式均导致了铁催化剂活性位的掩盖。费托合成是结构敏感型反应,催化剂的结晶度和晶体结构对催化性能具有重要的影响。催化剂的结晶度顺序与催化剂稳定性顺序相一致,说明较高的结晶度和完整的活性相晶型不仅可以增加催化剂的机械强度,还可防止活性组分在高温下发生熔结而影响其使用寿命。这也证明在费托合成反应中,提高催化剂的结晶度有利于促进催化剂的活性和稳定性。
3 结论
本文考察4种制备方法(并流共沉淀,正加共沉淀,沉淀-浸渍K,沉淀-浸渍K、SiO2)对草酸亚铁制备的100Fe/4Cu/4K/16SiO2铁催化剂的结构、物化性质以及费托合成催化性能的影响。
(1)通过SEM 和BET 的表征结果得知,制备方法影响催化剂的结晶方式,使催化剂具有不同外貌结构和织构性质。催化剂比表面积的大小顺序为:沉淀-浸渍K>并流共沉淀>正加共沉淀>沉淀-浸渍K、SiO2。沉淀-浸渍K 法能明显提高催化剂的比表面积。
(2)XRD 和H2-TPR 分析可知4 种方法制备的铁催化剂的主要物相均为Fe2O3和Fe3O4的混合物,催化剂在H2中的低温还原性顺序为:并流共沉淀>沉淀-浸渍K>正加共沉淀>沉淀-浸渍K、SiO2。这主要是由于助剂与Fe 相的结合方式不同导致了催化剂还原程度的难易。
(3)并流共沉淀制备的催化剂由于具有较好的结晶度和还原性,助剂与活性相结合程度更为均匀,使得催化剂具有最佳的活性和稳定性。且催化剂表现出较高的烃类产物收率和低碳烯烃选择性;正加共沉淀制备的催化剂表现出较低的甲烷选择性;沉淀-浸渍K方法制备的催化剂对C5+的选择性最高。
(4)对4种方法制备催化剂的液相产物和固相产物进行碳数分析发现,并流共沉淀所得的液相产物中汽油组分的含量最高,且C4+重质烃产物中,C4~C18质量分数高达91.0%;正加共沉淀的液相产物中煤油组分的选择性较高;而沉淀-浸渍K 的液相产物中柴油组分含量较多。
猜你喜欢
空间科学学报(2020年3期)2020-07-24
丝绸(2020年6期)2020-06-23
CHINESE JOURNAL OF AERONAUTICS(2017年5期)2017-11-17
雷达学报(2017年1期)2017-05-17
安徽医科大学学报(2015年9期)2015-12-16
江苏农业科学(2015年1期)2015-04-17
科技创新导报(2014年34期)2015-01-13
