
超声预处理对大豆蛋白聚集体结构和乳化特性的影响
2020-06-29 01:18陈凡凡王中江
农业机械学报 2020年6期
李 杨 陈凡凡 王中江 滕 飞
(东北农业大学食品学院, 哈尔滨 150030)
0 引言
大豆分离蛋白(Soy protein isolate,SPI)是一种全价蛋白[1],常作为乳化剂应用于乳制品、肉制品和面制品中,通过降低水和油的表面张力、水和空气的表面张力,达到改善食品风味、颜色、质地和储存稳定性以及提高产品质量的目的[2-3]。在加热、杀菌、干燥等加工过程中,蛋白质极易受到温度的影响,进而发生构象改变,造成蛋白功能活性的衰减,而热聚集体的形成是导致蛋白丧失某些生物和功能特性(如蛋白的溶解性、乳化活性和乳化稳定性)的重要原因[4-6]。热聚集体的形成主要包括两个阶段:分子解聚成亚基形式,随后亚基逐渐去折叠,此时分子内部疏水残基逐渐暴露,表面巯基含量增高;当这些分子累积的能力足够彼此接近以实现聚合物形成时,去折叠的亚基通过疏水相互作用迅速聚集形成聚集体[7]。近年来,国内外学者对热诱导聚集领域进行了大量的研究,考察了热诱导聚集对蛋白结构和功能性质的影响[8],并尝试通过各种物理方法来解聚热聚集体,以达到提高蛋白质功能特性的目的[9],但对于调控热诱导可溶性聚集体形成的研究较少。
超声作为一种非热物理加工技术,能产生局部的空化效应,改变分子间氢键、范德华力、疏水性等维持蛋白质结构的作用力,促进蛋白构象改变,进而引起蛋白乳化特性的改变,因此引起广泛关注。超声技术应用范围较广[10],蛋白经过超声处理后,其溶解度发生变化[11],且超声对蛋白的作用分为两种:一种是蛋白结构预修饰,以增强功能性,然后应用于生产;另一种是将超声直接作用于生产体系,增强蛋白与其他物质结合的稳定性。目前,关于超声改性提高蛋白特性的研究较多,研究发现,超声处理可提高米糠蛋白溶解性和乳化性[12]、破坏黑豆蛋白质分子的内部疏水作用、解聚和重组黑豆蛋白结构[13],而适当的超声功率和超声时间会使黑豆蛋白的粒径最小化、电位绝对值最大化、蛋白表面疏水性提高[14]。研究表明超声可用于改性蛋白增加蛋白功能特性和结构稳定性,但经超声处理的蛋白进行热处理后,其结构和功能特性变化的相关研究却未见报道。蛋白自身特性可直接影响加工生产中的构象改变和功能特性发挥,本文将超声技术作用于大豆蛋白,然后将处理后的蛋白进行常规加工的热诱导处理,利用超声稳定蛋白结构的特性,抑制蛋白因过热处理而发生的聚集,进而提高蛋白的功能特性,以解决热诱导引起蛋白功能衰减的难题。
本文以超声处理为预处理技术,并对超声后的蛋白进行热诱导,探究超声处理对蛋白结构特性(微观结构、粒径分布、官能团、二三级结构、电位、疏水性)和乳化特性(乳化性和乳化稳定性)的影响,分析蛋白结构的改变与蛋白乳化特性的关系。研究超声功率和超声时间对蛋白热聚集的抑制效果,并探究其原因,为解决热效应造成蛋白热聚集而使蛋白功能衰退的难题提供方法和理论。
1 材料与方法
1.1 材料与试剂
大豆分离蛋白(纯度91.12%),山东禹王集团;2,4-二硝基苯肼,天津博迪化工股份有限公司;三氯乙酸,天津市致远化学试剂有限公司;叠氮化钠,山东浩中化工有限公司;乙酸乙酯,天津市光复精细化工研究所;β-巯基乙醇、过硫酸铵,Geniview公司;2-硝基苯甲酸(DTNB)、乙二胺四乙酸(EDTA),天津市博迪化工有限公司;邻苯二甲醛(OPA),广州奈姆塔贸易有限公司;8-苯胺萘磺-1-酸盐(ANS),上海将来实业股份有限公司;尿素(Urea)试剂,武汉博士康生物工程有限公司;十二烷基硫酸钠(SDS),索莱宝生物科技有限公司。其他试剂均为分析纯等级。
1.2 仪器与设备
Biosafer 650-92型超声波细胞破碎仪,江苏赛飞信息技术有限公司;HH-6型数显水浴锅,山东爱博科技贸易有限公司;LW-1600FC型紫外可见分光光度计,上海菁华科技仪器有限公司;Zetasizer Nano ZSP型马尔文纳米粒度电位仪,马尔文仪器公司;F-4500型荧光分光光度计,日本HITACHI公司;MAGNA-IR560型傅里叶变换红外光谱,美国尼高力公司;Ultra-Turrax T25型高速分散器,德国IKA公司;ALPHA 1-4 LSC型冷冻干燥机,德国Christ公司;PHSJ-4A型实验室pH计,上海仪电科学仪器股份有限公司;DK-98-1型电热恒温水浴锅,天津市泰斯特仪器有限公司;XW-80A型旋涡混合器,上海青浦沪西仪器厂;GL10M型立式高速冷冻离心机主机,北京新诺立华仪器有限公司;FJ200-S型数显高速均质机,邢台浩诚科技开发有限公司。
1.3 蛋白超声预处理和加热诱导处理
利用0.01 mol/L、pH值7.4的磷酸盐缓冲溶液将大豆分离蛋白(SPI)溶液(含0.5 mg/mL NaN3)质量浓度调配为10 mg/mL,超声(对照;6 min, 200 W;6 min,400 W;6 min,600 W; 12 min,600 W;24 min,600 W)处理之后转移至100℃恒温箱中20 min,诱导大豆蛋白发生热聚集。将热聚集液体于低温离心机中9 000 r/min离心20 min,弃去沉淀物后留存上清液经过24 h冷冻干燥后即可得到6种可溶性蛋白样品,再加上可溶性大豆分离蛋白(SSPI)(蛋白溶液经离心冻干所制),即7个样品,分别记为SSPI、STSPI、6 min 200 W-STSPI、6 min 400 W-STSPI、6 min 600 W-STSPI、12 min 600 W-STSPI和24 min 600 W-STSPI。
1.4 蛋白的微观结构
通过扫描电镜对样品的表面形貌进行观察。将样品均匀平摊在贴有导电胶的样品台上,喷金处理,观察成像时加速电压为20 kV,观测倍数为10 000。
1.5 粒径分布测定
将可溶性蛋白溶于水中配成质量浓度为0.05 g/mL的溶液,搅拌均匀后,缓慢加入测量池中,用仪器测量其粒径电位和蛋白质分散度指数(PDI)。
1.6 浊度的测定
参照文献[15]的方法,并稍作修改。将样品溶于去离子水中配制成所需的浓度,在室温(20℃)下磁力搅拌60 min。分光光度计在600 nm下测定其吸光度。以去离子水作空白,测定其吸光度A,浊度计算公式为
(1)
式中V——稀释倍数
I——光程距离,取0.01 m
1.7 游离氨基含量的测定
参照文献[16]的方法,并稍作修改。400 mg OPA试剂充分溶解于1 mL甲醇溶液中,而后依次向溶液中加入预先配置的质量浓度为200 g/L的SDS溶液2.5 mL及质量浓度为0.1 mol/L的硼酸溶液25 mL,继而转移至通风橱内加入100 μL的β-巯基乙醇,最后将溶液用蒸馏水定容至50 mL,制备成OPA溶液用于后续检测分析,量取OPA试剂4 mL与200 μL蛋白样品充分混匀后进行35℃水浴处理2 min,以蒸馏水空白组为对照在340 nm处测定吸光度。
1.8 游离巯基和二硫键含量的测定
参照文献[17]的方法,并稍作修改。称取400 mg的DTNB,加入Tris-Gly(氨丁三醇-甘氨酸)缓冲液定容至100 mL,配成Ellman试剂。分别称取2 mg样品溶解于2 mL的Tris-Gly缓冲液(pH值8.0)和0.02 mL的Ellman试剂。测定时溶液振荡快速混合后在25℃下保温反应15 min,用分光光度计测定其在412 nm处的吸光度,以不加Ellman试剂为空白。将样品用磷酸盐缓冲液配制成0.5 mL质量浓度为5 mg/mL的蛋白质溶液,置于10 mL塑料离心管中,加入2.5 mL含8 mol/L尿素的Tris-Gly(10.4 g Tris,6.9 g Gly,每1 L加1.2 g EDTA,pH值8.0),每隔1.5 min加入20 μL DTNB(0.004 g DTNB用Tris-Gly溶解定容至1 mL,避光),反应25 min,立即在412 nm处测吸光度。对照组不加蛋白质溶液,其他处理方法相同。根据标准曲线(y=0.050 2x-0.000 9,R2=0.999 4)计算出蛋白质质量浓度ρ。摩尔消光系数取13 600 L/(mol·cm),巯基质量摩尔浓度计算公式为
(2)
式中S——巯基质量摩尔浓度,μmol/g
A412——加Ellman试剂时412 nm处样品的吸光度
D——稀释系数
C——样品蛋白最终质量浓度,mg/mL
1.9 羰基含量的测定
参照文献[18]的方法,并稍作修改。将蛋白用去离子水配制为5 mg/mL的蛋白溶液,以双缩脲指示剂法测定蛋白质溶液的质量浓度(y=0.241 2x+0.013 5,R2=0.997)。在367 nm处用2, 4-二硝基苯肼比色法进行比色,每毫克蛋白质羰基衍生物的摩尔数通过摩尔消光系数22 000 L/(mol·cm)进行计算,公式为
(3)
式中G——蛋白羰基质量摩尔浓度,nmol/mg
n——稀释因子
A367——367 nm处吸光度
E——蛋白质溶液的质量浓度,mg/mL
1.10红外光谱分析
参照文献[19]的方法,并稍作修改。将冻干样品置于干燥器内充分干燥,称取1 mg样品于100 mg溴化钾中混匀,在玛瑙研埚中研磨并用压片器压片,于红外光谱仪中测定吸收光谱。测量条件:波数范围为4 000~400 cm-1,分辨率4 cm-1,波数精度0.01 cm-1,扫描次数64次,环境温度25℃。
1.11内源性光谱分析
参照文献[20]的方法,并稍作修改。称取一定量样品于5 mmol磷酸缓冲液(pH值 7.0)配成质量浓度为0.001 g/mL的溶液,取适量样品置于荧光分光光度计中测量。测量条件:激发波长290 nm,发射波长300~400 nm,狭缝宽均为5 nm,电压为700 mV。
1.12Zeta电位的测定
参照文献[21]的测定方法,并稍作修改。采用Zetasizer Nano ZSP型马尔文纳米粒度电位仪对蛋白溶液的Zeta电位进行测定。将蛋白样品分散到50 mmol/L、pH值7.0的磷酸盐缓冲液中,配成质量分数为0.2%的蛋白溶液,取样量为3 mL,测定温度为25℃。
1.13 表面疏水性指数H0测定
参照文献[22]的ANS荧光探针法测定样品表面疏水性的方法,并稍作修改。用0.1 mol/L的中性磷酸盐缓冲溶液稀释,10 000 r/min高速离心处理0.5 h除去沉淀物,以Lowery法分析测试上清液中蛋白浓度,通过磷酸盐缓冲液的逐步稀释,调控蛋白溶液质量浓度至0.05~0.4 mg/mL,取40 μL浓度为8 mmol/L的ANS溶液滴加至不同浓度的蛋白溶液4 mL,经振荡混匀后静置3 min,在荧光分光光度计下进行荧光强度测试,测试条件为:激发波长λex=390 nm,发射波长λem=468 nm,扫描夹缝宽度设置为5 nm,扫描速度设置为10 nm/s。将荧光强度与蛋白质量浓度作线性图,初始段的斜率计为样品的表面疏水性指数。
1.14乳化性和乳化稳定性的测定
参照文献[23]的方法,并稍作修改。在测试管中分别加入15 mL质量浓度0.001 g/mL蛋白质溶液和5 mL玉米油,乳液经高速均质机(24 000 r/min)处理1 min后,从测试管底部取出50 μL乳液,用0.001 g/mL的SDS溶液稀释100倍后,于500 nm比色。乳化性指数EAI和乳化稳定性指数ESI的计算公式为
(4)
(5)
式中EAI——乳化性指数,m2/g
ESI——乳化稳定性指数,min
F——稀释因子,取100
A0—— 0 min时的吸光度
A30——30 min时的吸光度
H——蛋白质量浓度,g/mL
φ——光程,取0.01 m
θ——油相质量分数,取25%
1.15数据处理和分析
每个实验均进行3次重复平行实验,利用SPSS Statistics 22软件对数据进行ANOVA差异显著性分析,以P<0.05为显著性差异。采用Origin 9.1软件、Peak Fit 4.12软件等进行数据分析、图表处理及图谱分析处理。
2 结果与分析
2.1 可溶性蛋白的微观结构变化
由图1箭头指出的孔洞结构可知,SPI在经过热处理后由片状结构聚集成网状结构;随着超声功率的增大,USTSPI微观图中箭头所指的网状结构由松散杂乱逐渐变得致密且孔洞均匀;但当超声功率不变,随着超声时间的增大,超声后热可溶性蛋白的结构逐渐由致密均匀逐渐变得松散。这表明超声处理可以影响蛋白的聚集效果,进而影响蛋白的微观结构[24]。

图1 超声预处理对蛋白微观结构的影响Fig.1 Effects of ultrasonic pretreatment on microstructure of protein
2.2 热处理对预处理蛋白粒径的影响
由图2和表1可知,与SSPI相比,STSPI的平均粒径、PDI和浊度均显著增大,且粒径分布图由原来的单峰变成了三峰;与STSPI相比,经过超声预处理的蛋白在热诱导时粒径、PDI和浊度显著降低,随着超声功率的增大,其粒径分布图逐渐左移,且STSPI最右侧峰消失,表中平均粒径、浊度和PDI逐渐减小;与6 min 600 W-STSPI相比,随着超声时间的增大,粒径分布图逐渐右移,平均粒径、PDI和浊度呈现增大的趋势。
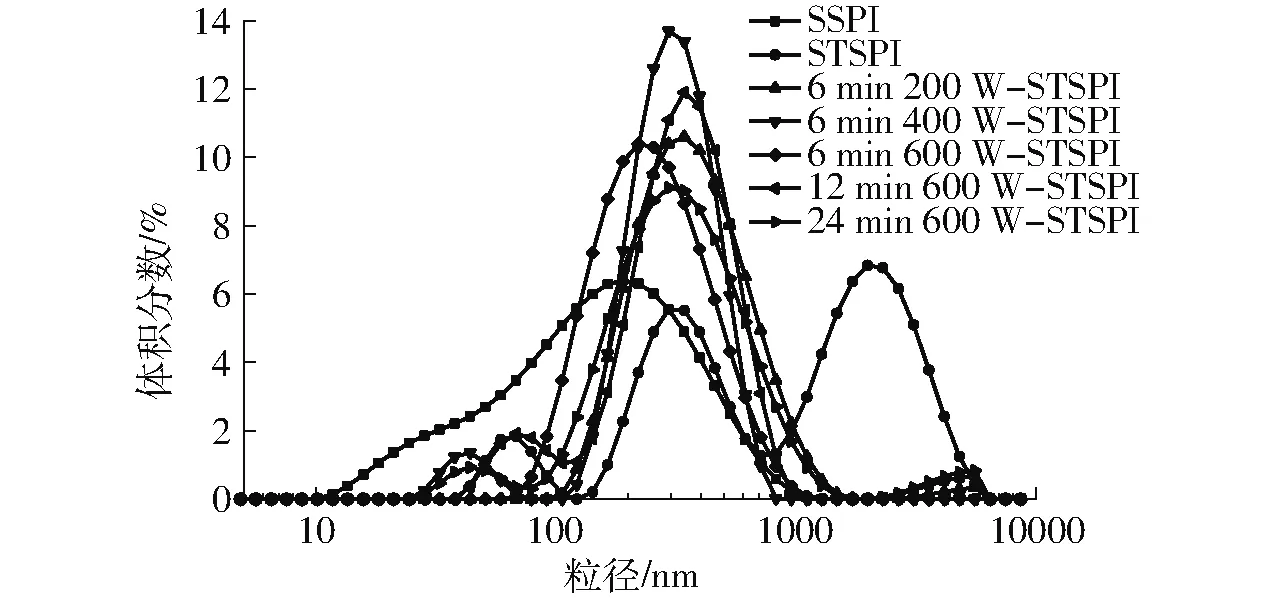
图2 超声预处理对蛋白粒径分布的影响Fig.2 Effect of ultrasonic pretreatment on protein particle size distribution
加热处理可使蛋白先解聚后聚集,导致粒径、PDI和浊度增大。而超声预处理使蛋白发生一定程度的疏水性基团暴露,增强颗粒间的静电排斥力,从而提高蛋白质分散体系的稳定性[14]。超声功率越大,蛋白疏水性基团暴露得越明显,分子间斥力越大,抑制热聚集体形成的效果越明显[25]。而超声时间过长会导致蛋白分子在疏水作用力的情况下发生聚合,溶液稳定性变差,从而降低了抑制热聚集体形成的效果,并促进小分子聚集体向中等大小聚集体转变。粒径分布图可在一定程度上反映蛋白粒度的分布状况,而PDI 则反映粒径分布范围,而溶液浊度可以反映蛋白质的聚集程度,可从侧面衡量蛋白质相互作用大小及聚集体的相对尺寸。图2中超声预处理后峰图较STSPI左移,且大颗粒峰图消失,颗粒分布较为集中。从表1可知,经过超声预处理的蛋白PDI处于0.25~0.35之间,较SSPI的0.52和STSPI的0.58小,经过超声预处理的蛋白浊度也显著降低。粒径、PDI和浊度的变化趋势说明超声预处理在一定程度抑制了蛋白热聚集,且超声功率和时间的不同对热诱导聚集抑制程度不同。
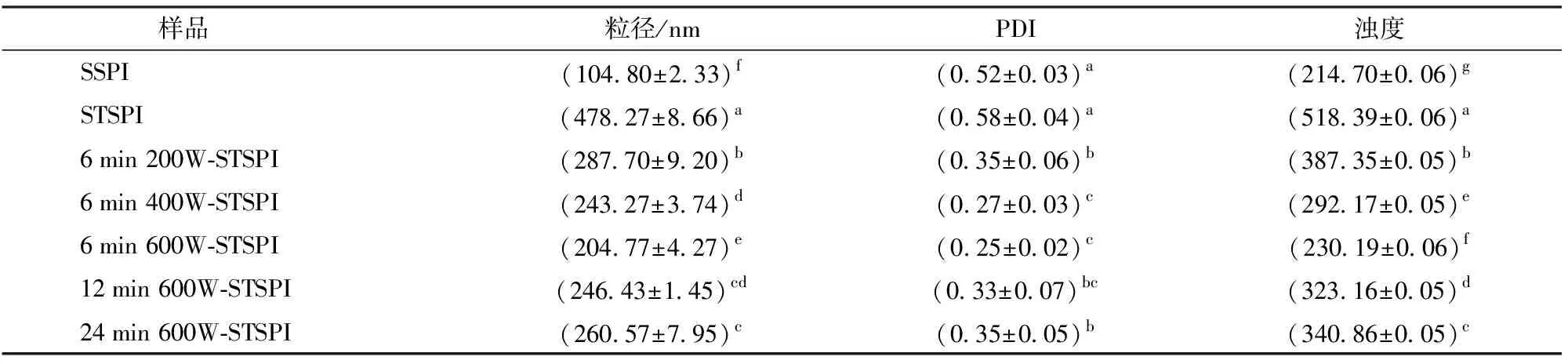
表1 超声预处理对蛋白结构的影响Tab.1 Effect of ultrasonic pretreatment on protein structure
注:同列中相同字母表示数据差异不显著(P>0.05),不同则表示差异显著(P<0.05),下同。
2.3 可溶性蛋白游离氨基和羰基含量的测定
蛋白结构在受热展开的过程中,埋藏在分子内部的氨基侧链暴露并很快被氧化生成羰基结构,氨基含量和羰基含量在一定程度上可用来表征蛋白结构的展开程度和氧化程度[26]。巯基基团(—SH)是蛋白中重要的功能基团,其含量变化可反映蛋白质的变性程度,对其功能性质的发挥具有很重要的作用[27]。如表2所示,与可溶性SPI相比,可溶性TSPI的游离氨基、游离巯基和总巯基含量减少,而羰基和二硫键含量增多;与STSPI相比,随着超声功率的增大,其游离氨基和巯基呈现升高趋势,而羰基含量和二硫键含量呈现减小趋势;与6 min 600 W-STSPI相比,随着超声时间的增大,游离氨基和巯基含量呈现降低趋势,而羰基含量先增大后减小,二硫键含量呈现升高趋势。
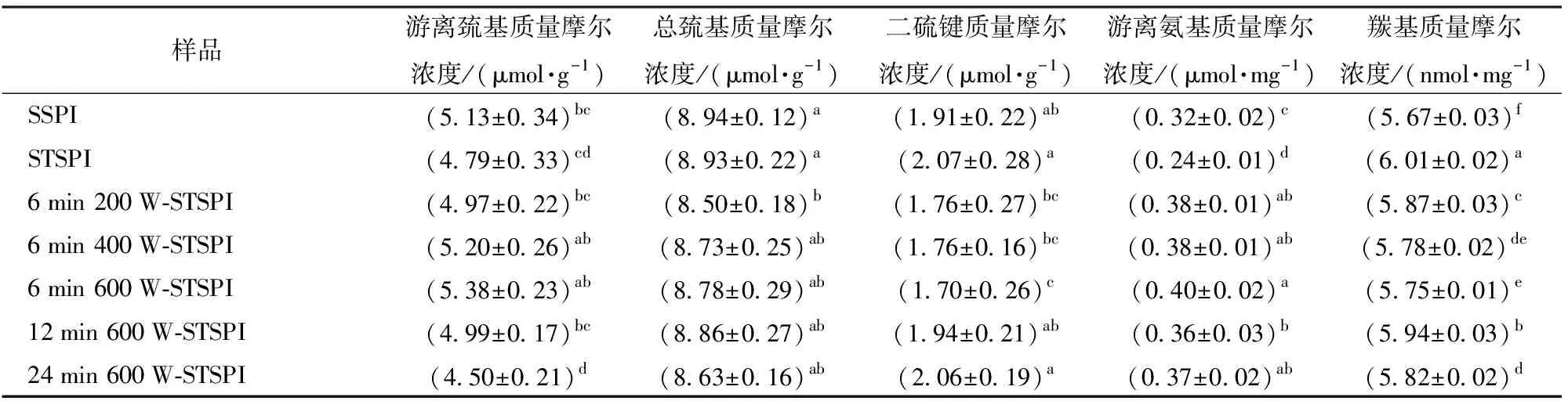
表2 游离氨基、巯基、二硫键和羰基含量Tab.2 Free amino, sulfhydryl, disulfide and carbonyl contents
蛋白溶液在进行热处理时,当蛋白聚集程度高于解聚时,巯基基团部分转换为二硫键,部分被重新掩藏,导致巯基含量降低,二硫键含量增加。蛋白结构在展开过程中产生的部分氨基因氧化生成新的羰基基团[26,28],羰基含量增多。已有研究证实,超声波的空化效应和剪切力可将大聚集体解聚转化为小聚集体,提高蛋白表面疏水性,增大巯基的暴露量,破坏分子间二硫键形成游离巯基[25],增加蛋白质表面负电荷,增强颗粒间静电排斥,并提高蛋白质分散体的稳定性[14],抑制蛋白聚集,进而减少羰基量。当超声功率为600 W和超声时间为6 min时疏水性基团的暴露量达到最大值,超声功率和时间持续增大,存在过量的自由基,促进羰基的形成[29]。蛋白之间非共价相互作用推动蛋白颗粒聚集,游离巯基和游离氨基减少,二硫键含量增多。
2.4 可溶性蛋白二级结构组分含量测定
傅里叶变换红外光谱是表征蛋白质二级结构的主要技术方法之一。热处理可断开蛋白α-螺旋结构中的氢键,使α-螺旋结构减少[30],β-转角结构和β-折叠结构(β1+β2)含量增多。而反向平行的折叠结构(β1)通常在聚集的蛋白质分子间形成[31],在一定程度上可表征蛋白的聚集程度。如图3和表3所示,与SSPI相比,STSPI的β1结构增多,α-螺旋和γ-无规则卷曲结构均减少;随着超声功率的增大,β1呈现减小趋势,α-螺旋和γ-无规则卷曲结构增多;与6 min 600 W-STSPI相比,随着超声时间的增大,β1增多,α-螺旋、β-转角和γ-无规则卷曲结构均减少。
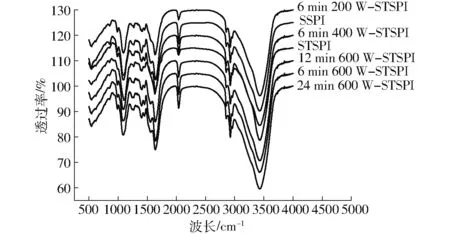
图3 蛋白的红外光谱图Fig.3 Infrared spectrum of protein
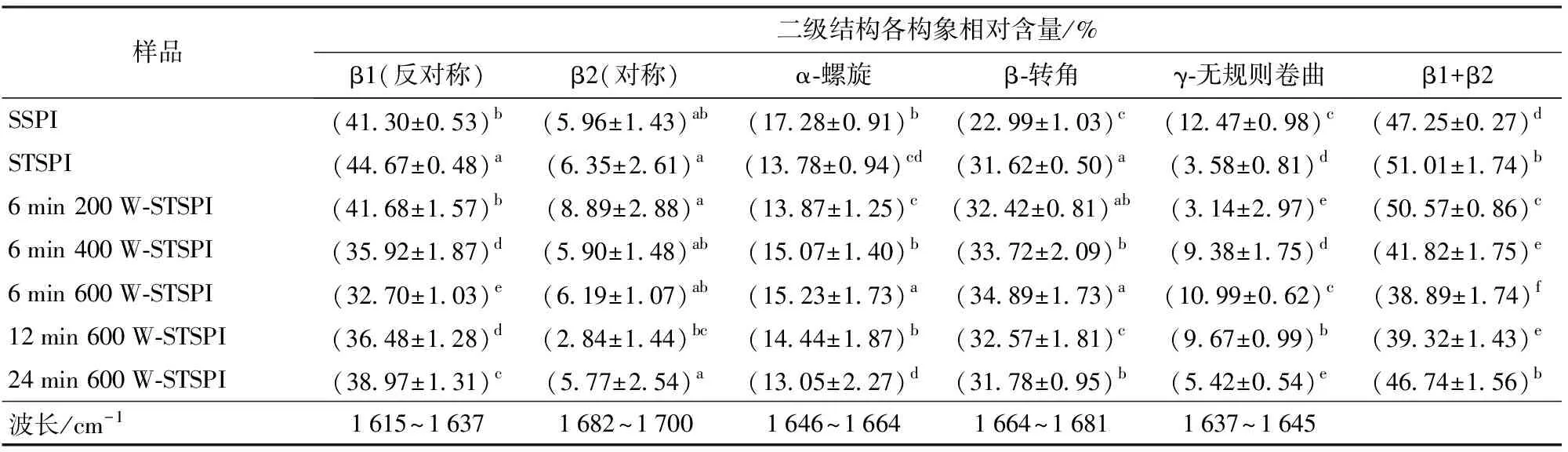
表3 超声预处理对蛋白二级结构的影响Tab.3 Effect of ultrasonic pretreatment on secondary structure of protein
研究表明,超声功率和超声时间的增大可能增加了蛋白质上的负表面电荷,而颗粒的有效表面电荷主要决定了它们的分散和聚集[32],增强颗粒间静电排斥,并提高蛋白质的稳定性[14]。与STSPI相比,超声后再经热诱导的可溶性蛋白β-折叠(尤其是β1)结构减少,α-螺旋、β-转角结构增多,可能与超声使蛋白表面电荷增多抑制蛋白受热聚集有关[25]。当超声条件为600 W、6 min时抑制蛋白聚集的能力达到最大,β1蛋白结构达到最小值。当超声时间进一步增大时,β-折叠结构增加[14],同时伴随着聚集体中α-螺旋结构减少,抑制聚集的能力减弱,这可能是局部氨基酸序列之间和部分分子之间相互作用被破坏所致[33]。这表明适当的超声预处理可在一定程度上抑制蛋白发生热聚集,这与之前的研究结果一致。
2.5 蛋白三级结构测定
蛋白质三级结构的变化可以通过内源性荧光的最大吸收波长的变化来确定[34]。由图4可知,与SSPI相比,STSPI的最大吸收波长出现蓝移现象;超声预处理后再经过热处理得到的可溶性蛋白,与STSPI相比,随着超声功率的增大,蛋白最大吸收波长出现不同程度红移;与6 min 600 W-STSPI相比,随着超声时间的增大,蛋白最大吸收波长出现蓝移。加热诱导蛋白聚集时,随着温度的增大,蛋白结构发生聚集,发色基团被掩藏,最大吸收波长发生蓝移[18]。随着超声功率增大,蛋白发生的红移现象可能与超声使蛋白结构柔性打开和伸展,暴露出疏水性基团和带电粒子,稳定的蛋白结构抑制蛋白进一步发生聚集有关,使蛋白暴露的发色基团较STSPI多,进而发生红移[35]。当超声功率和超声时间为600 W和6 min时,疏水性基团达到稳定值,蛋白溶液状态达到一个稳定态临界值。但随着超声时间的进一步增加,较高的疏水相互作用促使蛋白聚集,最大吸收波长较6 min 600 W-STSPI出现蓝移。
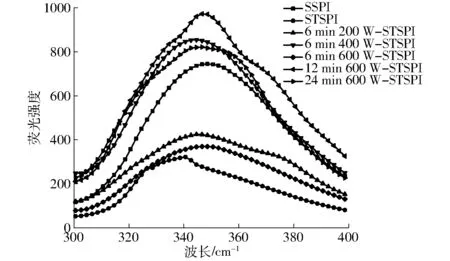
图4 超声预处理对蛋白三级结构的影响Fig.4 Effect of ultrasonic pretreatment on tertiary structure of protein
2.6 蛋白表面疏水性的测定
Zeta电位通常被用来表征溶液体系的稳定性,绝对值越大溶液越稳定,而表面疏水性变化侧面反映了大豆蛋白的结构变化[8],且对蛋白质的稳定性、构象和功能至关重要。由表4可知,与SSPI相比,STSPI的表面疏水性指数和电位绝对值增大;与STSPI相比,超声预处理蛋白电位绝对值整体减小,且随着超声功率的增大,蛋白的表面疏水性指数和电位绝对值呈现增大趋势;与6 min 600 W-STSPI相比,随着超声时间的增大,蛋白的电位绝对值呈现减小趋势,表面疏水性指数先增大后减小,且在6 min 600 W-STSPI电位绝对值达到最大值,在12 min 600 W-STSPI表面疏水性指数达到最大值。

表4 超声预处理对蛋白电位和表面疏水性的影响Tab.4 Effect of ultrasonic pretreatment on protein potential values and surface hydrophobicity
热处理可以打开蛋白结构,诱导蛋白质分子内部疏水残基和带电基团暴露,电位绝对值增大,而蛋白疏水相互作用的增大可促进蛋白发生聚集,导致蛋白表面疏水性降低[36],表中STSPI的表面疏水性高于SSPI可能与蛋白聚集方式发生变化导致部分疏水性基团仍处于暴露态有关。超声预处理导致电位绝对值整体减小,侧面反映了超声抑制聚集的作用。随着超声功率增大,诱导蛋白质子去折叠,从而导致最初在分子内部的疏水基团数量增加并暴露于极性周围环境[14],增大空气和水的界面面积,干扰分子间氢键和疏水相互作用[24],同时游离官能团和带电粒子数量的增多,增大了溶液中的静电作用力,进而增加溶液稳态,抑制了蛋白热诱导聚集现象,导致表面疏水性和电位绝对值增大[14]。且当处理功率和时间达到600 W、6 min时抑制能力达到最大,蛋白溶液状态达到一个稳定态临界值。随着超声时间的进一步增加,蛋白的抑制能力减弱,高功率超声波处理可能会增加蛋白结合程度,进而降低蛋白分散体稳定性,导致电位绝对值和表面疏水性减小[37]。
2.7 蛋白乳化性的测定
蛋白质分子因具有疏水和亲水的基团,从而能够分别作用于油相和水相,起到乳化剂的作用。蛋白质的热聚集会影响蛋白质分子的结构,进而影响蛋白质的乳化性。如图5(图中同一参数不同字母表示差异显著)所示,与SSPI相比,STSPI的乳化性指数和乳化稳定性指数降低;超声预处理后再经过热处理得到的可溶性蛋白,与STSPI相比,随着超声功率的增大,蛋白的乳化性和乳化稳定性呈现增大趋势;与6 min 600 W-STSPI相比,随着超声时间的增大,蛋白的乳化性和乳化稳定性呈现减小趋势,且在6 min、600 W时乳化性和乳化稳定性达到最大值。
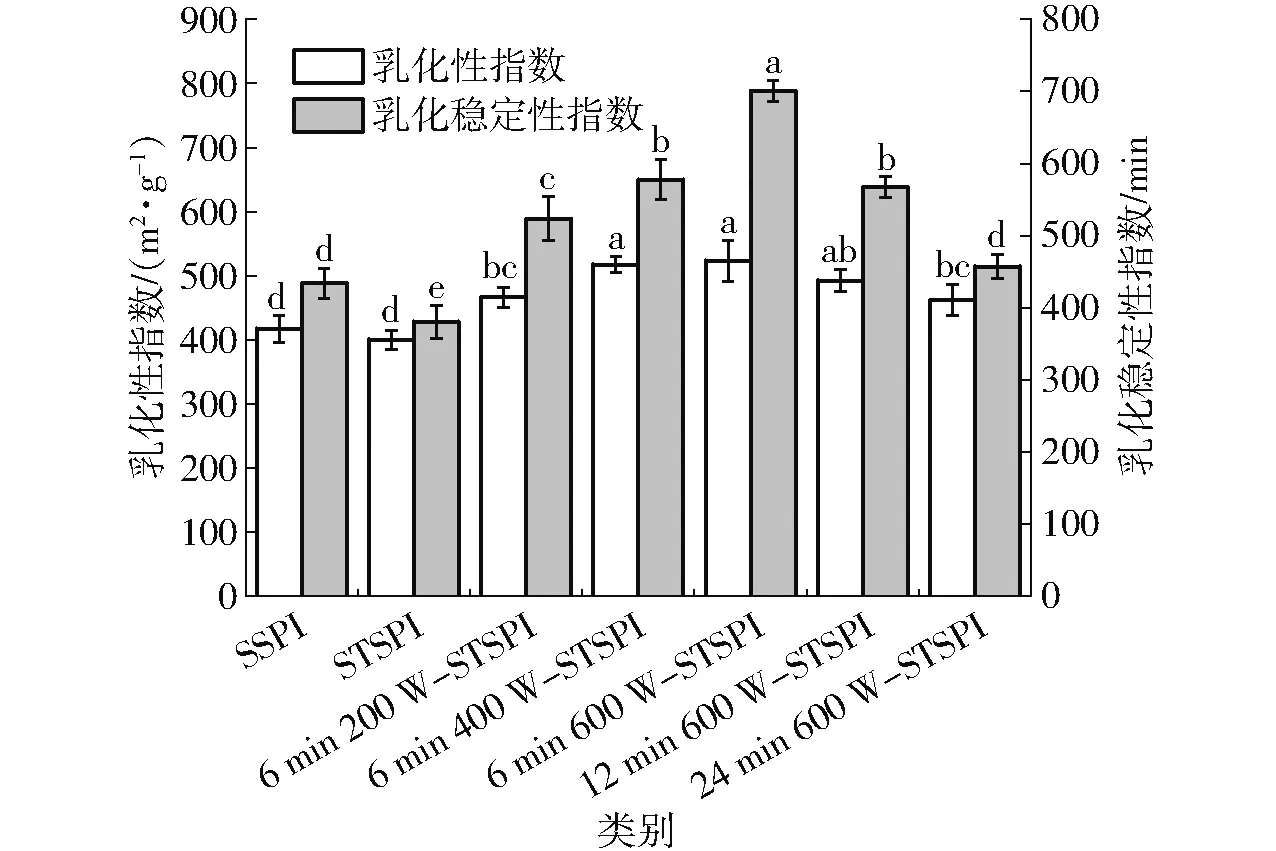
图5 超声预处理对蛋白乳化特性的影响Fig.5 Effect of ultrasonic pretreatment on protein emulsifying properties
热诱导降低了蛋白质分子疏水性指数/亲水性指数比值,蛋白质分子柔韧性降低,在油水界面不能迅速展开,进而降低了大豆蛋白的乳化性。超声功率和超声时间的增大可减小蛋白的粒径和增大蛋白的比表面积,增加分子间静电斥力和疏水相互作用,提高蛋白溶液稳定性,抑制蛋白聚集,提高乳液界面吸附蛋白能力,进而增强乳化性和乳化稳定性[38-39]。当超声时间和功率增大到6 min、600 W时,蛋白的疏水性基团的暴露量达到临界值。随着超声时间的增大,蛋白质分子内部疏水相互作用的破坏增加和蛋白质分子运动的加速使蛋白逐渐发生聚集,乳液界面上大分子颗粒蛋白极易被小分子蛋白置换,乳液界面张力增大,乳液发生絮凝,造成乳化性和乳液稳定性下降。
3 结论
(1)与SSPI相比,STSPI的平均粒径、PDI和浊度分别升高了373.47 nm、0.06和303.69;官能团中二硫键和羰基质量摩尔浓度分别升高了0.16 μmol/g和0.34 nmol/mg,游离巯基和氨基质量摩尔浓度分别降低0.34 μmol/mg和0.08 μmol/g;二级结构中α-螺旋结构减少,β-转角结构增加,β1结构相对含量提高了3.37个百分点;乳化性和乳化稳定性指数分别降低了16.77 m2/g和60.03 min,表面疏水性指数提高了113.21。这表明热聚集体可改变蛋白结构,并降低蛋白的乳化特性。
(2)与STSPI相比,经过超声预处理(6 min、 600 W)的蛋白在进行热处理后,其平均粒径、PDI、电位绝对值和浊度显著降低,而α-螺旋、β-转角结构、表面疏水性、乳化性和乳化稳定性提高,微观结构变得均匀致密。这表明适当的超声预处理可抑制因蛋白聚集而导致蛋白构象发生改变,进而防止蛋白因热聚集导致蛋白乳化特性的降低,这为解决因加热引起蛋白乳化特性衰减问题提供了方法和理论。
猜你喜欢
中国化妆品(2020年6期)2020-07-22
商品与质量(2019年31期)2019-11-28
意林·全彩Color(2018年9期)2018-10-12
筑路机械与施工机械化(2017年6期)2017-07-10
分析科学学报(2016年2期)2016-10-15
药学研究(2015年11期)2015-12-19
分析化学(2015年10期)2015-11-03
筑路机械与施工机械化(2015年11期)2015-07-01
食品工业科技(2014年23期)2014-03-11
食品工业科技(2014年13期)2014-03-11
