
八角茴香渗漉法的工艺优化及莽草酸的薄层色谱鉴别
2020-06-28 12:59:26周邦华龙杰凤王小羽汤承浩李本鹏
凯里学院学报 2020年3期
周邦华 ,龙杰凤,王小羽,汤承浩,谭 荣,李本鹏,王 翔,2*
(1.凯里学院,贵州凯里 556011;2.凯里学院黔东南民族药综合利用工程技术研究中心,贵州凯里 556011;3.沈阳药科大学,辽宁沈阳 117000)
八角茴香(Illicium verum)为木兰科八角属植物的干燥成熟果实,为著名的调味香料,果皮、种子、叶都含芳香油,是制造化妆品、甜香酒、啤酒和食品工业的重要原料[1-2].八角茴香除用作香料外,还用于治疗消化不良、胃痛、小儿氙气以及面部麻痹等[3].研究表明,其主要药效成分莽草酸及其衍生物通过影响花生四烯酸代谢,抑制血小板聚集和抑制动、静脉血栓及脑血栓形成,并具有抗炎、镇痛、抗肿瘤、抗血栓、抗脑缺血等作用[4-5].而且还是合成抗禽流感药物“达菲”的关键原料,可作为抗病毒和抗癌药物中间体在医药、食品等行业广泛应用[6].
八角茴香果实一般含莽草酸3%~10%,目前,文献报道提取莽草酸的方法有超声-微波协助提取法、水蒸气蒸馏法、超临界萃取法、溶剂回流提取法等[7-8],而相较于传统的渗漉法提取分离技术,不难发现,以上提取方法相对耗能,成本较高,往往因加热等因素会造成部分成分结构的破坏而影响药效.值得一提的是,笔者搜索知网等大型数据库发现,利用渗漉法这一传统溶剂提取法提取八角茴香中的莽草酸并进行薄层鉴别及纯化分离的文献报道实属罕见.因此,本文采用渗漉法、UV 法和薄层色谱鉴别法,通过单因素试验和L9(34)正交设计试验考察乙醇浓度、料液比、提取时间、药材粒度、渗漉速度与渗漉次数对八角茴香提取工艺的影响,建立八角茴香渗漉液中莽草酸含量测定及定性鉴别的方法,以期为后续开展八角茴香中莽草酸的进一步分离纯化奠定基础.
1 药材、试剂及仪器
1.1 药材及试剂
本实验所用药材为八角茴香(Illicium verum)(批号:20181101,安徽四全药业销售有限公司),经凯里学院王翔教授鉴定为木兰科八角属植物的干燥成熟果实,实验样品保存在凯里学院民族药标本馆.
本实验所用试剂有莽草酸对照品(批号:111835-201503,中国食品药品检定研究院,纯度99.7%);乙酸乙酯(西陇科学股份有限公司),三氯甲烷(重庆川东化工有限公司),甲酸(成都金山化学试剂有限公司),乙醇和甲醇(深圳市科天化玻仪器有限公司),正己烷、石油醚和丙酮(天津市东丽区天大化学试剂厂),以上均为分析纯.
1.2 实验仪器
紫外分光光度计(上海仪电分析仪器有限公司,L5 型),磁力搅拌器(上海豫康科教仪器设备有限公司,CL-2 型恒温加热型),电子天平(奥豪斯仪器(上海)有限公司,AR224CN 型),恒温水浴锅(常州天瑞仪器有限公司,HH-8 数显恒温型),洁拓超声波清洗机(深圳市洁拓超声波清洗设备有限公司),电热鼓风干燥箱(北京中兴信业仪器有限公司,10-1A型)等.
2 实验方法与结果
2.1 八角茴香药材处理及水分测定
取八角茴香药材饮片适量,筛选,粉碎过筛,分别制成3 批样品S1、S2、S3,置于干燥器备用.按照ChP2015(通则0832)第二法(烘干法)测定[1].见表1,结果八角茴香水分在12.15%~13.28%,平均值12.97%,低于相关规定的13.00%,表明药材保存完好,符合实验要求.
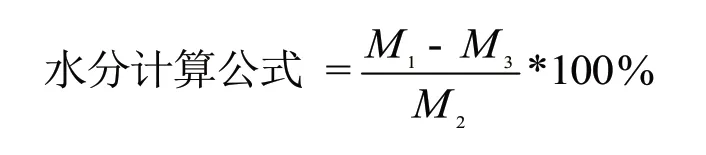
式中:M1为干燥前样品+称量瓶重量(g),M2为样品重量(g),M3为干燥后样品+称量瓶重量(g).
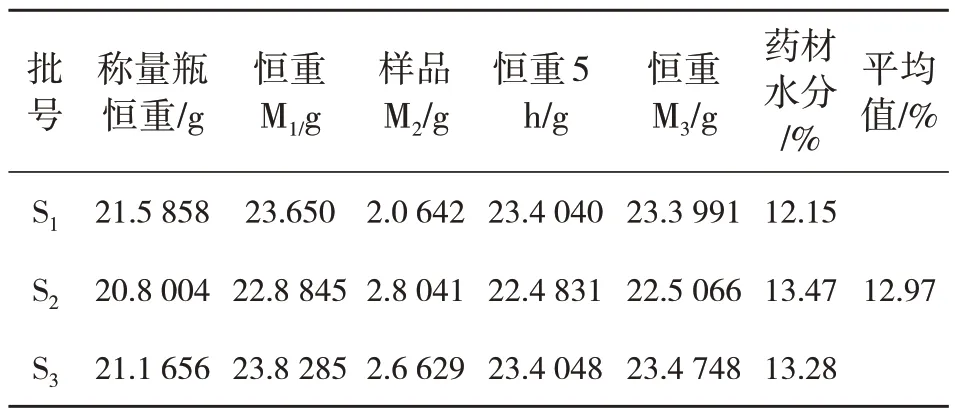
表1 八角茴香水分测定结果
2.2 干膏得率测定
按照ChP2015(通则0832)法测定[1].取供试品溶液25 mL,置干燥至恒重的蒸发皿中水浴蒸干,在100~105 ℃干燥3 h,移置干燥器中,放冷30 min,精密称定,再在上述温度干燥1 h,放冷,称重,至连续2 次称重的差异不超过5 mg 为止.根据减失的重量,计算样品中的含水量.

2.3 八角茴香渗漉液中莽草酸含量测定
2.3.1 供试品溶液制备
参照2015 版《中国药典》八角茴香饮片鉴别项下供试品溶液制备方法并适当调整[1,9-11],即精密吸取渗漉提取液10 mL,加入10 mL 石油醚-乙酸乙酯(1:1)溶液,超声处理10 min,77 ℃挥去上层液,加甲醇定容至25 mL,即得供试品溶液.
2.3.2 对照品溶液制备
精密称取10.0 mg 莽草酸对照品,置于50 mL容量瓶中,用甲醇溶解至刻度,即得0.2 mg/mL的对照品溶液[12].
2.3.3 标准曲线绘制
精密称取0.2 g 莽草酸对照品,用95 %乙醇定容于100 mL量瓶中,汲取1、3、5、7、9 mL置于10 mL量瓶中,再分别加95%乙醇定容至刻度,以空白试剂作参比,在227 nm 波长[11,13]处照紫外分光光度法测定莽草酸系列浓度的吸光度值,以莽草酸浓度(C)为横坐标,吸光度(A)值为纵坐标,绘制莽草酸的标准曲线图1.结果回归方程为y=0.5446x+0.1383,r=0.9992,表明在0.2~1.4 mg/mL 时,莽草酸浓度(C)对吸光度(A)有良好的线性关系.
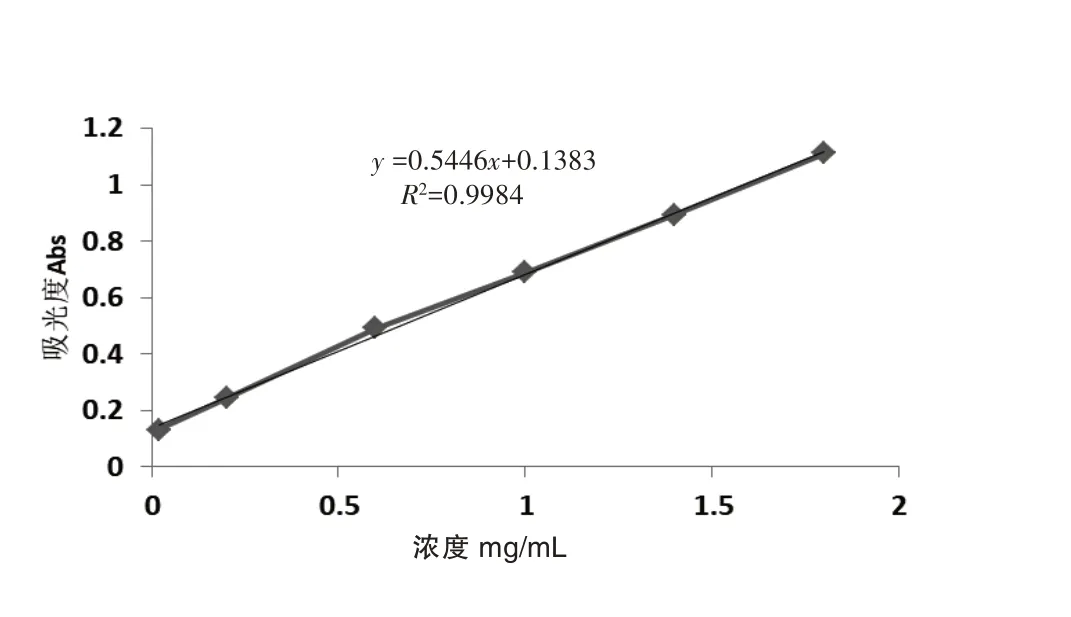
图1 莽草酸的标准曲线图
2.3.4 精密度考察
精密量取2.3.2 项下同一浓度莽草酸对照品溶液,照2.3.3项下测定方法,连续进样6次,比较吸光度.结果相对标准偏差RSD 为1.5%,小于2%,表明所用仪器精密度良好.
2.3.5 重复性考察
精密量取同批八角茴香渗漉液6份,按2.3.1项下方法制备供试品溶液,照2.3.3项下测定方法重复进行测定,比较吸光度的RSD.结果相对标准偏差RSD为2.1%,表明重复性良好,满足分析要求.
2.3.6 稳定性考察
精密量取八角茴香渗漉液,按2.3.1 项下制备供试品溶液,分别于0、2、4、6、8、10、12 h 时,照2.3.3 项下测定方法测定,比较吸光度,结果相对标准偏差RSD 为1.03%,小于2%,表明样品供试液在12 h内稳定性良好,符合要求.
2.3.7 加样回收率试验
精密量取莽草酸已知浓度为0.5793 mg/mL 的八角茴香渗漉供试品溶液5 mL,6 份,各加70%乙醇定容于10 mL 量瓶中,分别按样品中莽草酸含量的50%、100%、150%加入莽草酸标准品,混匀,按2.3.1 项下制备供试品溶液,依法测其吸光度并计算加样回收率和RSD 值,见表2.结果样品平均加样回收率为98.89%,RSD 为1.74%,表明在规定范围内,符合要求.
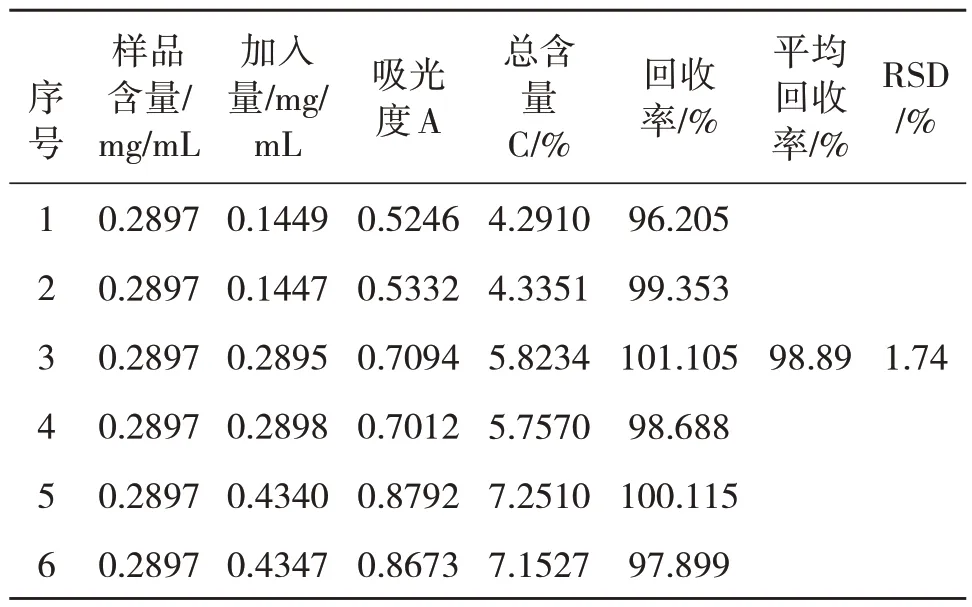
表2 加样回收率试验结果
2.3.8 含量测定
称取八角茴香样品约5 g,平行3 次,照优化的工艺条件制得渗滤液,照2.3.1 项下制备方法制备供试品溶液,测定吸光度,带入2.3.3 项下回归方程,计算样品中莽草酸含量,结果见表3,测得样品中莽草酸平均含量为9.40%.
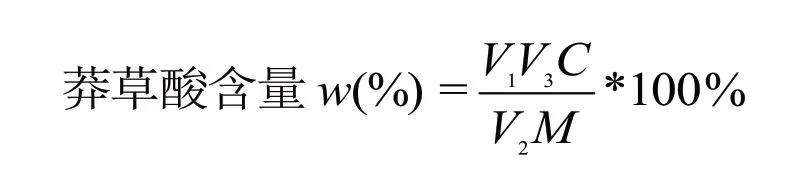
其中W为样品中莽草酸百分含量,V1为渗漉液总体积(mL),V2为从渗漉液总体积中取出的体积(mL),V3为待测液体积(mL),M为八角粉末取样量(mg),C为回归方程读取的质量浓度(mg/mL).
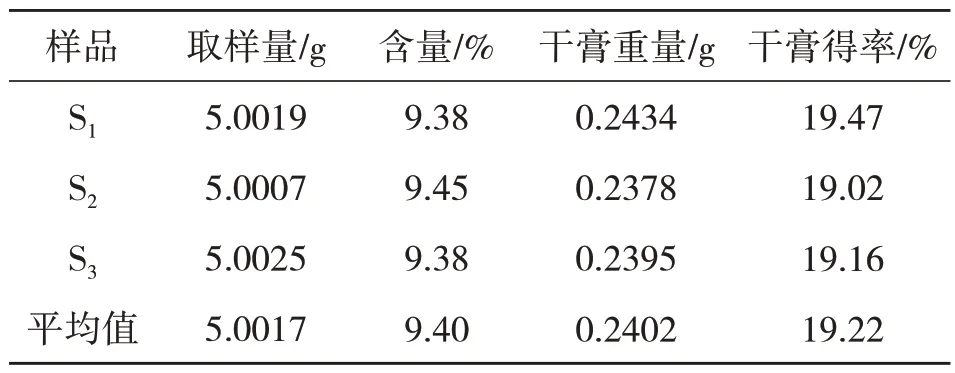
表3 含量测定结果
2.4 提取工艺研究
2.4.1 单因素实验
本实验根据各指标因素对提取工艺影响的贡献大小分配权重系数[14-16],即分别设定莽草酸和干膏得率的权重系数为0.6和0.4,采用综合评分法评分.综合评分=(W莽草酸含量/W莽草酸含量最大值)*0.6+(W干膏率/W干膏率最大值)*0.4.
2.4.1.1 乙醇浓度考察
保持其他因素不变,称取药材粉末(80目筛)5 g,在料液比为1:10 g/mL,浸泡时间12 h 时,30%、50%、70%、90%乙醇浓度对八角茴香莽草酸含量和干膏得率及综合评分的影响见图2,结果表明,相较于其他浓度,70%的乙醇浓度提取效果较好,故选择70%的乙醇溶液.
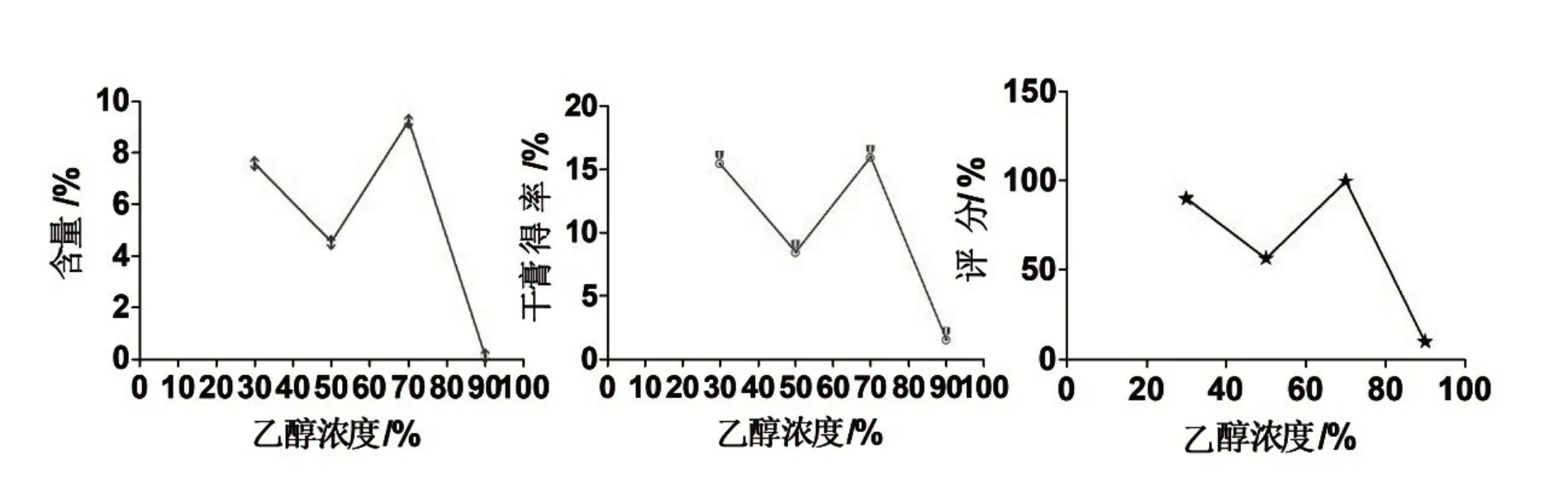
图2 乙醇浓度考察
2.4.1.2 料液比考察
保持其他因素不变,称取药材粉末(80 目筛)约5 g,浸泡时间12 h,乙醇浓度70%时,料液比为1:10、1:15、1:20、1:25、1:30、1:35 g/mL 对八角茴香莽草酸含量和干膏得率及综合评分的影响见图3,结果显示,相较于其他料液比,料液比1:10 g/mL时效果较好,选择料液比1:10 g/mL.
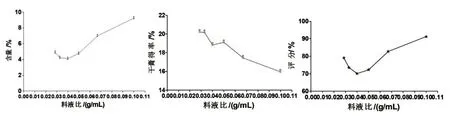
图3 料液比考察
2.4.1.3 浸泡时间考察
称取药材粉末(80 目筛)约5 g,乙醇浓度为70%,料液比为1:10 g/mL 时,保持其他因素不变,泡时间为6、12、18、24、30、36、42 h对八角茴香莽草酸含量和干膏得率及综合评分的影响见图4,由图知,浸泡时间为12 h 时效果较好,故选择浸泡时间为12 h.

图4 浸泡时间考察
2.4.1.4 粒度考察
称取药材粉末约5 g,乙醇浓度为70%,料液比为1:10 g/mL,浸泡时间12 h 时,保持其他因素不变,药材粒度(20 目、40 目、60 目、80 目、100 目)对八角茴香莽草酸含量和干膏得率及综合评分的影响如图5,由图分析,粒度为80目时效果较好,故选择80目.
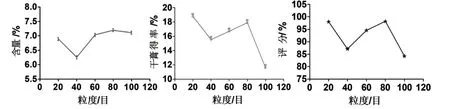
图5 粒度考察
2.4.1.5 提取次数考察
保持其他因素不变,称取药材粉末(80 目筛)约5 g,乙醇浓度为70%,料液比为1:10 g/mL,浸泡时间12 h 时,考察渗漉1、2、3 次对八角茴香莽草酸含量和干膏得率及综合评分的影响如图6,结果显示,渗漉数1次时的测定结果较好,故选择渗漉1次即可.
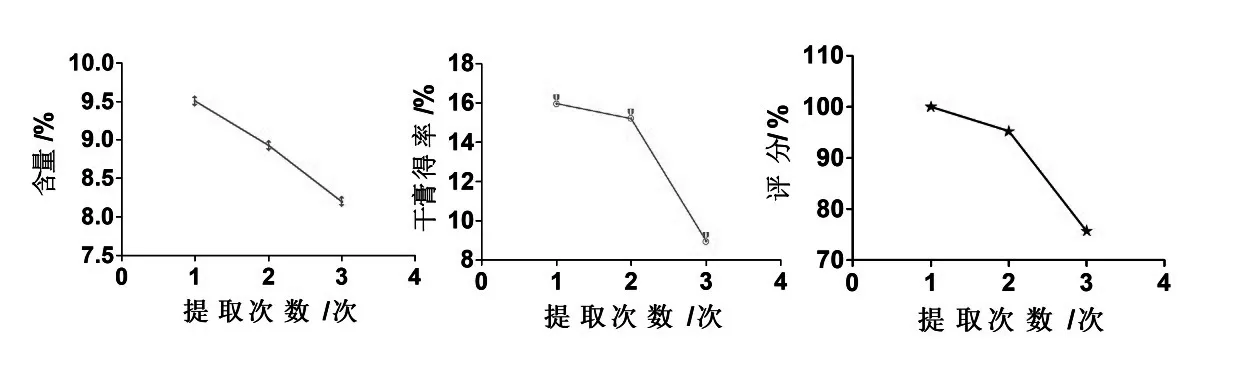
图6 提取次数考察
2.4.1.6 渗漉流速考察
称取药材粉末(80 目筛)约5 g,乙醇浓度为70%,料液比为1:10 g/mL,浸泡时间12 h 时,保持其他因素不变,渗漉流速1、2、3 mL/min 对八角茴香莽草酸含量和干膏得率及综合评分的影响见图7,结果显示在渗漉1 次的情况下,流速1 mL/min时,结果较优.故选择渗漉流速1 mL/min.
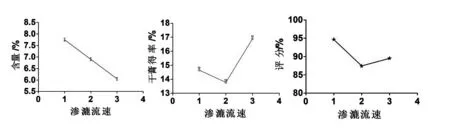
图7 渗漉流速考察
2.4.2 正交试验设计
参考文献[14-18]方法进行测定,在单因素试验基础上,以莽草酸含量及干膏率及综合评分为考察指标,选取浸泡时间(A)、料液比(B)、乙醇浓度(C)为考察因素,按L9(34)正交设计表进行渗漉法提取实验,因素水平见表4,正交实验安排及结果见表5,方差分析见6.

表4 因素-水平表
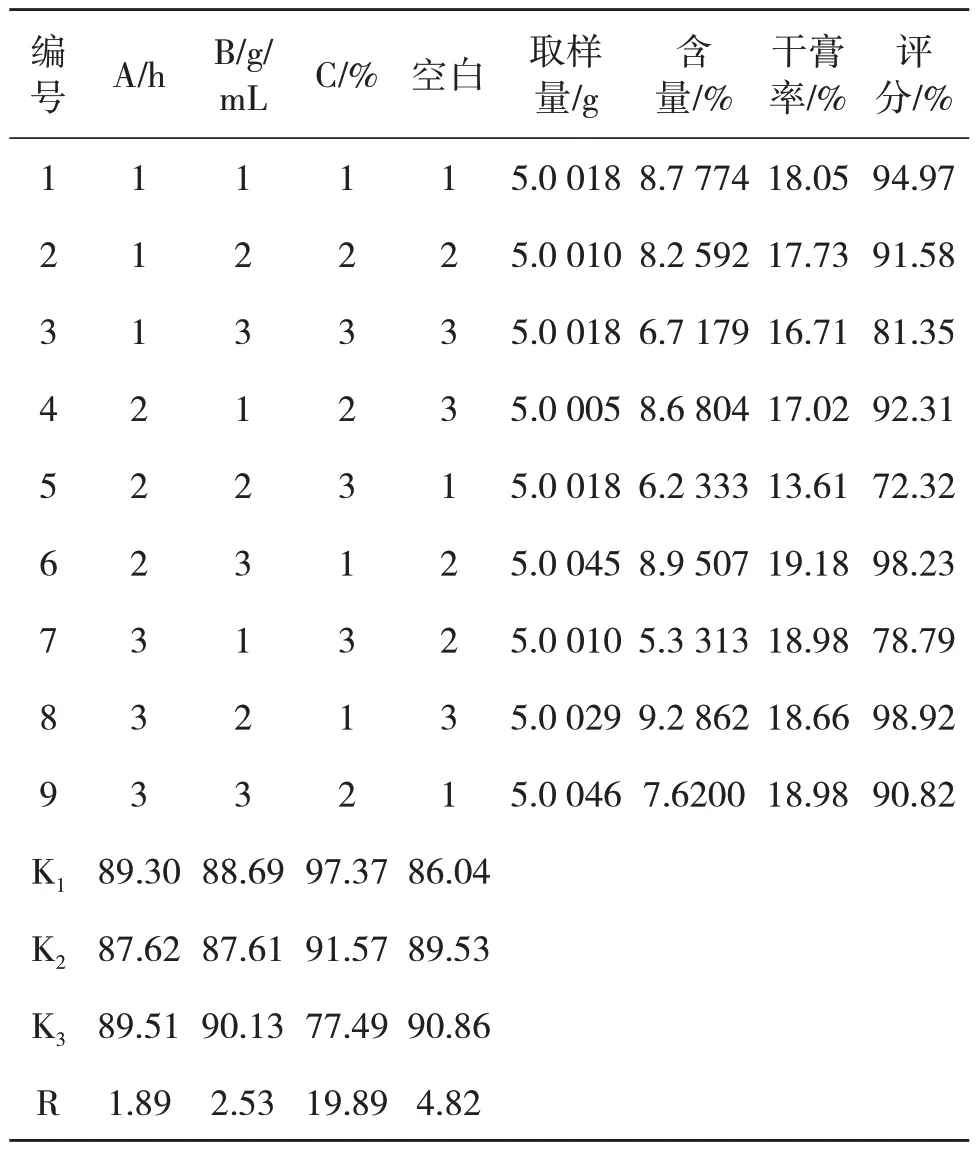
表5 正交实验设计安排与结果
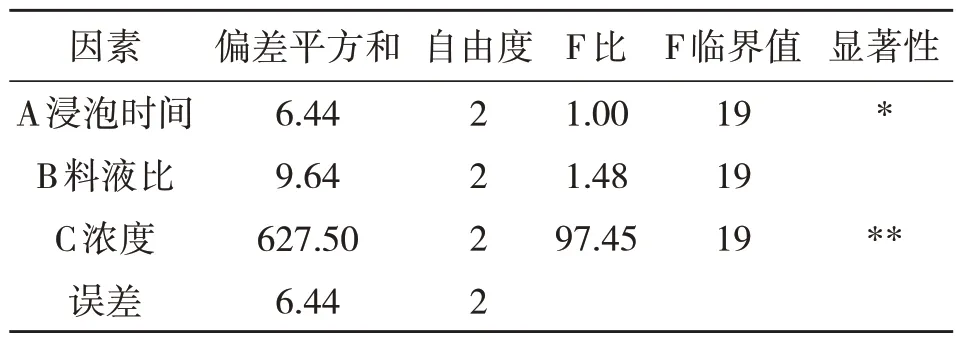
表6 方差分析结果
结果表明,由上表的直观分析和极差分析结果可以看出,极差RC>RB>RA,3 个因素对莽草酸提取率的影响依次为:乙醇浓度(C)>料液比(B)>浸泡时间(A),各因素中乙醇浓度的影响最为显著,料液比次之,浸泡时间影响较小,后两者的作用相当.在实验设计范围内,综合生产实际和单因素实验结果,评估因素水平取值,预测可能的最佳工艺条件为A2B2C1,即乙醇浓度60%、浸泡时间12 h,料液比1:15 g/mL.
2.4.3 验证实验
取80目八角茴香粉末3份(平行3份),约0.5 g,根据正交设计实验结论预测的工艺条件A2B2C1进行渗漉提取,照2.3.1项下制备供试品溶液,按2.3.3项下测定方法进行测定,结果见表7.结果表明:莽草酸平均含量为9.40%,高于正交试验表5 中的每一项实验结果,故A2B2C1条件即乙醇浓度60%、浸泡时间12 h,料液比1:15 g/mL 为渗漉法最佳提取工艺条件.
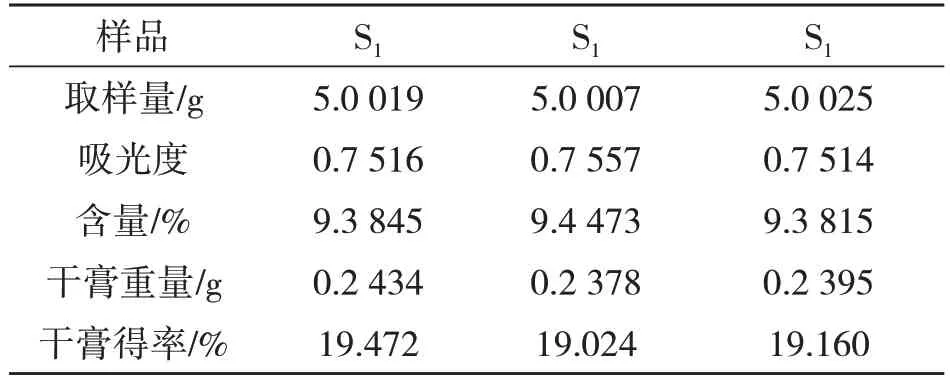
表7 验证实验结果表
2.5 八角茴香提取物中莽草酸的薄层色谱鉴别
2.5.1 供试品溶液制备
收集最优工艺条件下的渗漉液,滤过,60 ℃真空浓缩干燥,粉粹过筛(80 目),精密称取干膏粉末约0.5 g,参照2.3.1项下供试品溶液制备方法,制成20 mg/mL的供试品溶液.
2.5.2 对照品溶液制备
精密称取莽草酸对照品适量,参照2.3.2 项下对照品溶液制备方法,制成1 mg/mL 的对照品溶液.
2.5.3 薄层鉴别方法(参照文献[1,12]并适当调整展开剂系统)
2.5.3.1 薄层色谱点样量考察
分别精密吸取供试品、对照品各1、2、3、4、5、6µL点于同一种硅胶G 薄层板上,以三氯甲烷-甲醇-正己烷-甲酸(5:1.5:1:0.1)为展开剂展开,取出,晾干,喷1%的硫酸溶液的1%的高锰酸钾溶液,如图8.结果表明点样量为3µL、Rf 为0.56 时,可较好分离,故选择点样量为3µL.
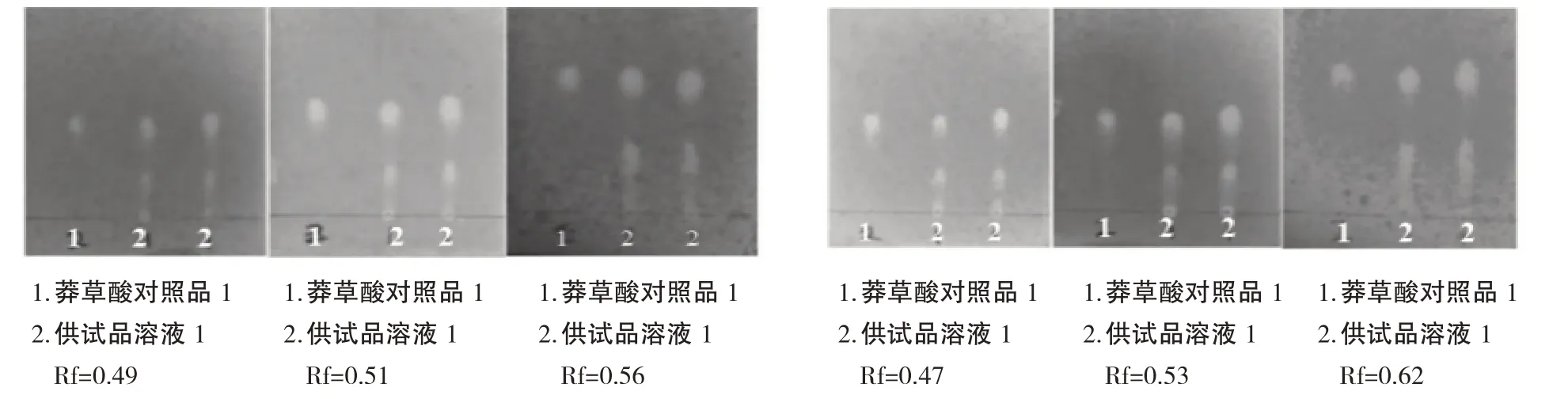
图8 薄层色谱点样量考察
2.5.3.2 薄层色谱温度考察
精密吸取供试品、对照品各3µL,分别在环境为5 ℃、16 ℃、20 ℃、室温、25 ℃、30 ℃下,保持其他条件不变照上述鉴别方法操作,结果如图9,显示温度为25 ℃,Rf 为0.46 时,分离效果较好,故选择温度为25 ℃.
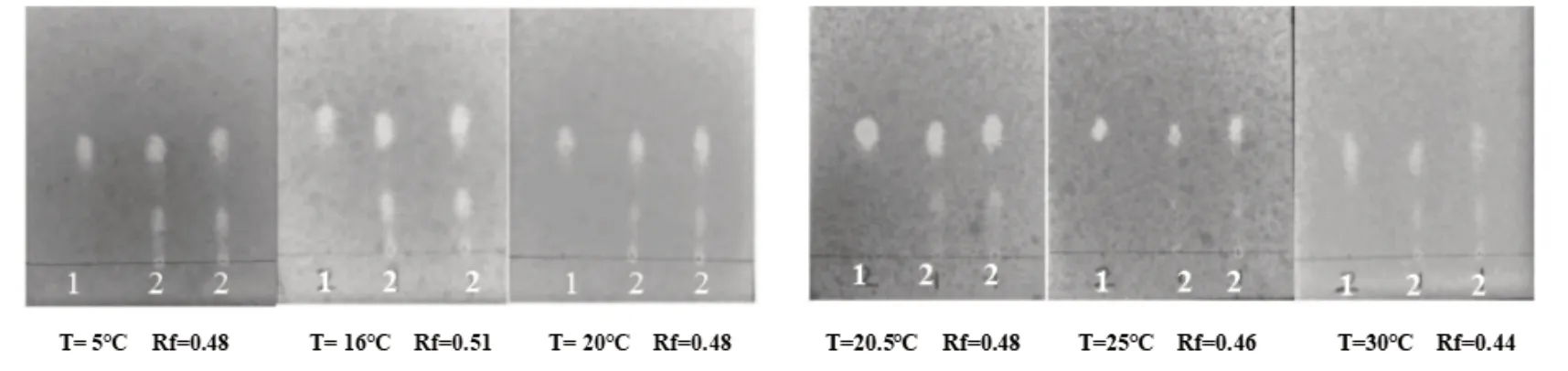
图9 薄层色谱温度考察
2.5.3.3 薄层色谱湿度考察
精密吸取供试品、对照品各3 µL,温度为25 ℃,保持其他条件不变照上述鉴别方法操作,20%、30%、40%、50%、60%、70%、80%的湿度环境下的结果如图10,提示湿度40%,Rf 为0.54 时,分离效果较好,故湿度选择40%.
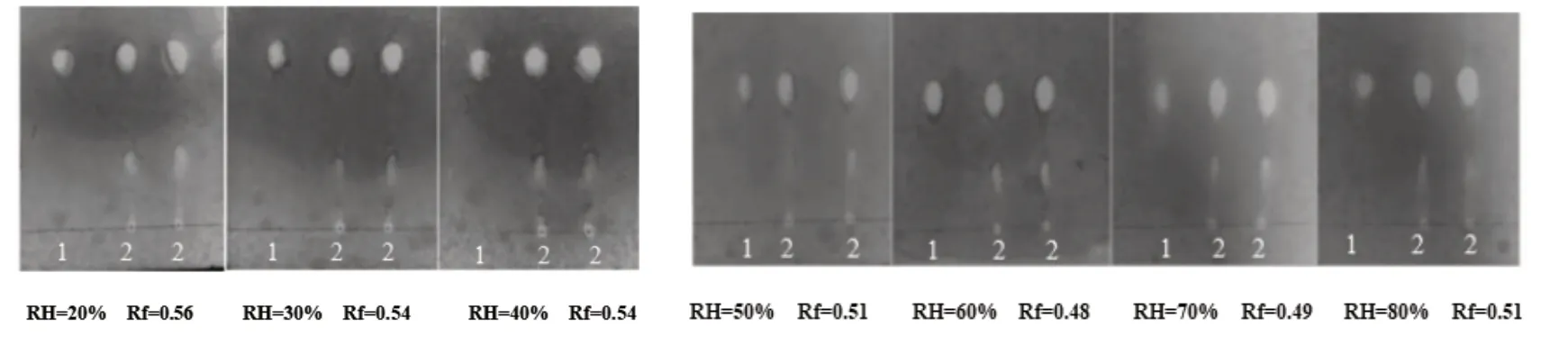
图10 薄层色谱湿度考察
2.5.3.4 薄层色谱板考察
在温度为25 ℃,湿度40%下,精密吸取供试品、对照品各3 µL 分别点于硅胶G 薄层板、硅胶GF254薄层板、自制薄层板和聚酰胺薄膜上,保持其他条件不变照上述鉴别方法操作,结果如图11,结明薄层板为硅胶GF254、Rf 为0.63 时效果较好,故选择硅胶GF254薄层板.
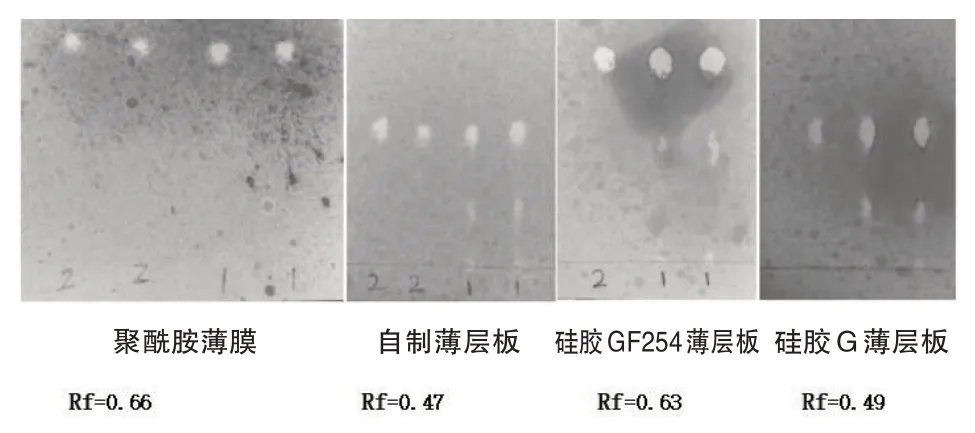
图11 薄层色谱板考察
2.5.3.5 薄层色谱展开剂考察
在温度为25 ℃,湿度40%时,精密吸取供试品、对照品各3 µL 分别以①三氯甲烷-甲醇-正己烷-甲酸(5:1.5:1:0.1)、②三氯甲烷-甲醇(6:4)、③石油醚-丙酮-乙酸乙酯(19:1:1)为展开剂,保持其他条件不变照上述鉴别方法操作,结果如图12,表明展开剂为①三氯甲烷-甲醇-正己烷-甲酸(5:1.5:1:0.1)、Rf 为0.52 时斑点分离清晰,故选择①为展开剂.
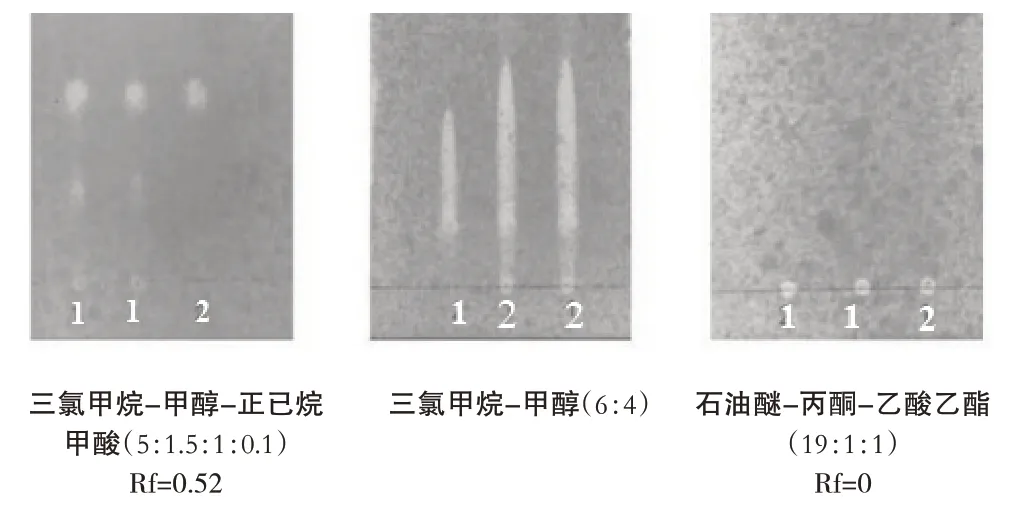
图12 薄层色谱展开剂考察
2.5.3.6 薄层色谱鉴别最终确定条件
参照薄层色谱法(附录ⅥB)试验,吸取供试品溶液和对照品溶液各3 µL,点于硅胶GF254薄层板上,在温度为25℃、湿度为40%的条件下,以三氯甲烷-甲醇-正己烷-甲酸(5:1.5:1:0.1)为展开剂展开,取出,晾干,喷以1%的硫酸溶液的1%的高锰酸钾溶液,供试品色谱中,在与对照品色谱相应的位置上,显相同颜色的斑点,莽草酸最佳条件下的薄层色谱如图13.
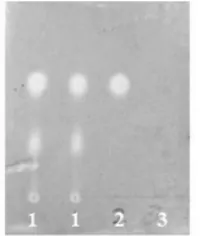
图13 莽草酸的薄层色谱图(1.八角茴香供试品溶液;2.莽草酸对照品溶液;3.空白溶液)
3 讨论
本实验单因素试验结果研究表明,当乙醇浓度为70%、料液比1:10 g/mL、粒度80 目、渗漉时间为12 h、提取次数为第1 次、渗漉流速1 mL/min(慢)时,结果最优;正交试验及验证试验研究结果表明,当乙醇浓度为60%、料液比1:15 g/mL、渗漉时间12 h 时,测得八角茴香中的莽草酸含量最高达9.45%,正交试验优化的八角茴香渗漉法提取工艺研究结果较好.
本设计完成了八角茴香水分、干膏得率、莽草酸含量的测定及提取物中莽草酸的鉴别研究,对精密度、重复性、稳定性和加样回收率等进行了方法学考察,在单因素试验和正交设计试验基础上优化渗漉法提取莽草酸工艺参数.该方法操作简便,工艺稳定,可为后期八角茴香中莽草酸的分离纯化研究提供实验参考.
猜你喜欢
中国粮油学报(2018年12期)2018-03-19 05:40:38
临床医药文献杂志(电子版)(2017年11期)2017-05-17 04:48:41
材料科学与工程学报(2016年4期)2017-01-15 13:35:34
饮食科学(2016年3期)2016-07-04 15:12:40
饮食科学(2016年3期)2016-07-04 15:12:27
海峡科技与产业(2016年3期)2016-05-17 04:32:14
广西林业科学(2016年2期)2016-03-20 05:53:43
分析测试学报(2015年4期)2016-01-13 06:18:29
云南中医学院学报(2015年3期)2015-07-31 18:09:28
中国合理用药探索(2014年11期)2014-03-11 20:30:20
