
细菌纤维素合成菌纳塔葡糖酸醋杆菌遗传操作方法的建立
2020-06-21 15:37谢文平鲍素敏刘权张鸿
安徽农业科学 2020年11期
谢文平 鲍素敏 刘权 张鸿

摘要 [目的]建立纳塔葡糖酸醋杆菌遗传操作系统,为采用基因工程方法改造该类菌株,提高纤维素产量,提升纤维素品质提供参考依据。[方法]通过优化培养基确定菌株电转化感受态制备条件,采用质粒或者线性化片段在不同电压下进行转化,优化最佳转化条件。[结果]采用添加了1%纤维素酶的C2培养基进行1次摇瓶转接,可有效制备纳塔葡糖酸醋杆菌电转化感受态;采用携带同源臂及Kan抗性基因的环状质粒或者线性化片段均实现了对纳塔葡糖酸醋杆菌葡萄糖脱氢酶基因(GDH)的同源双交换基因敲除;GDH基因的敲除降低了工程菌株发酵产纤维素膜过程的葡萄糖酸产生,提高了细菌纤维素的最终产量及品质。[结论]成功建立了纳塔葡糖酸醋杆菌遗传转化方法和基因敲除方法。
关键词 细菌纤维素;纳塔葡糖酸醋杆菌;电转化;基因敲除
中图分类号 S-3 文献标识码 A
文章编号 0517-6611(2020)11-0001-06
Abstract [Objective]Establishing a genetic operating method for Gluconacetobacter nataicola can provide reference for modifing the strains by genetic engineering,improving the yield and the quality of bacterial cellulose.[Method]The conditions for the preparation of competent cells were determined by the growth ability of the strain in different media,and the optimal transformation conditions were determined by positive clone obtained by transforming the plasmid or the linearized fragment at different electrotransformation voltages.[Result]The C2 medium supplemented with 1% cellulase was used for one time bottom transferring,which can effectively prepare the electroporation of G.nataicola.The homologous doubleexchange knockout of the glucose dehydrogenase gene (GDH) was achieved by transferring the circle plasmid or the linearized fragment carrying homologous arm of GDH and Kan resistance gene.Knockout of GDH gene reduced the production of gluconic acid in the process of fermentation,and improved the final yield and quality of bacterial cellulose membrane.[Conclusion]The genetic transformation method and gene knockout method of G.nataicola were successfully established.
Key words Bacterial cellulose;Gluconacetobacter nataicola;Electroporation;Gene knockout
細菌纤维素(bacterial cellulose,BC)是细菌利用葡萄糖合成的由β-1,4-糖苷键连接而成的纤维素。因其具有超高的纯度、高结晶性、高杨氏模量、优良的生物可降解性、高持水量和良好的生物相容性等特点,已作为新型生物可降解材料用于食品、化学工业和医学领域[1-2]。目前,细菌纤维素已经成功应用于烧伤敷料、退热贴、高端面膜等产品的基材而在各国实现了规模化生产[3-5]。
根据已有的报道,具有细菌纤维素合成功能的微生物包括葡糖酸醋杆菌属(Gluconacetobacter)、土壤杆菌属(Agrobacterium)、醋酸杆菌属(Acetobacter)、无色杆菌属(Achromobacter)、肠杆菌属(Enterobacter)等[3,6-7]。其中,葡糖醋酸菌属的木醋杆菌(G.xylinum)、巴氏醋杆菌(G.pasteurianus)、汉式醋杆菌(G.hansenis)、纳塔葡糖酸醋杆菌(G.nataicola)等种属的微生物都具有相对较强的纤维素合成能力[6]。
目前,对产纤维素菌种的研究主要集中在发酵条件的优化上,以提高纤维素合成能力[8-11]。采用基因工程方法进行育种具有更好的目标性和有效性,也是目前微生物育种常用的方法。但是由于细菌纤维素生产菌株为非模式生物,有关其遗传转化的方法研究很少。2006年,Chien等[12]采用广宿主质粒在木醋杆菌中表达了血红素蛋白,增强菌株的摄氧能力,提高菌株的细菌纤维素合成能力。2015年Kuo等[13]在木醋杆菌中采用同源重组的方法敲除了其中的GDH基因,提高了纤维素产量。2016年,Florea等[14]在G.rhaeticus(又称Komagataeibacter rhaeticus)中构建了一套遗传操作工具,用于在该菌株中进行外源基因的表达。但目前对于葡糖酸醋杆菌属的其他种属,如纳塔葡糖酸醋杆菌的遗传操作鲜有报道。
该研究以纳塔葡糖酸醋杆菌为对象,通过摸索优化感受态制备条件和转化条件,实现对该种菌的基因敲除操作,以期为优化纳塔葡糖酸醋杆菌在工业上的应用奠定基础。
1 材料与方法
1.1 材料
1.1.1 菌株和质粒。
纳塔葡糖酸醋杆菌HEC-004,是一株快速、高产细菌纤维素的工业化生产菌株,已保藏于中国普通微生物管理中心,保藏号:CGMCC No.11588。大肠杆菌DH5a,载体构建用菌株。pMD18-T载体购于宝生物工程(大连)有限公司,pET-28a质粒为Novagen公司产品。
1.1.2 试剂和设备。
PCR用的高保真DNA聚合酶PrimeSTAR Max DNA Polymerase、普通DNA聚合酶EmeraldAmp PCR Master Mix (2× Premix)、连接酶Solution I、 QuickCut限制性内切酶、DNA marker、质粒提取及DNA凝胶回收试剂盒均购于宝生物工程(大连)有限公司。纤维素酶,硫酸卡那霉素(Kan)和氨苄青霉素(Amp)购于生工生物工程(上海)股份有限公司。其余化学试剂为国产分析级试剂。
主要设备:PCR仪(Biometric),凝胶成像系统(BioRad),电穿孔仪器(Bio-Rad,型号165-2100)。
1.1.3 培养基。
大肠杆菌所用LB培养基:酵母粉5.0 g/L、蛋白胨10.0 g/L、氯化钠10.0 g/L。根据需要,在大肠杆菌质粒构建过程中加Kan 至终浓度50 mg/L或Amp至终浓度100 mg/L。纳塔葡糖酸醋杆菌所用的培养基有3种(表1),其中固体培养基加入2%琼脂。7#培养基用于菌种的基础培养,C2培养基用于菌株发酵产纤维素膜。
1.2 方法
1.2.1 纳塔葡糖酸醋杆菌对Kan和Amp抗生素的耐受性测试。
将纳塔葡糖酸醋杆菌HEC-004菌株接到7#液体培养基中,28 ℃ 振荡培养2 d后,取菌液稀释100倍,取50 μL分别涂布到Kan浓度为 0、5、25、50、150、100 mg/L的7#培养基固体平板上;按照上述相同方法,取50 μL分别涂布到Amp浓度为 0、25、50、100、150、200 mg/L的7#培养基固体平板上。上述平板置于28 ℃ 培养箱中培养,每天观察平板上菌落生长状况,连续观察7 d。
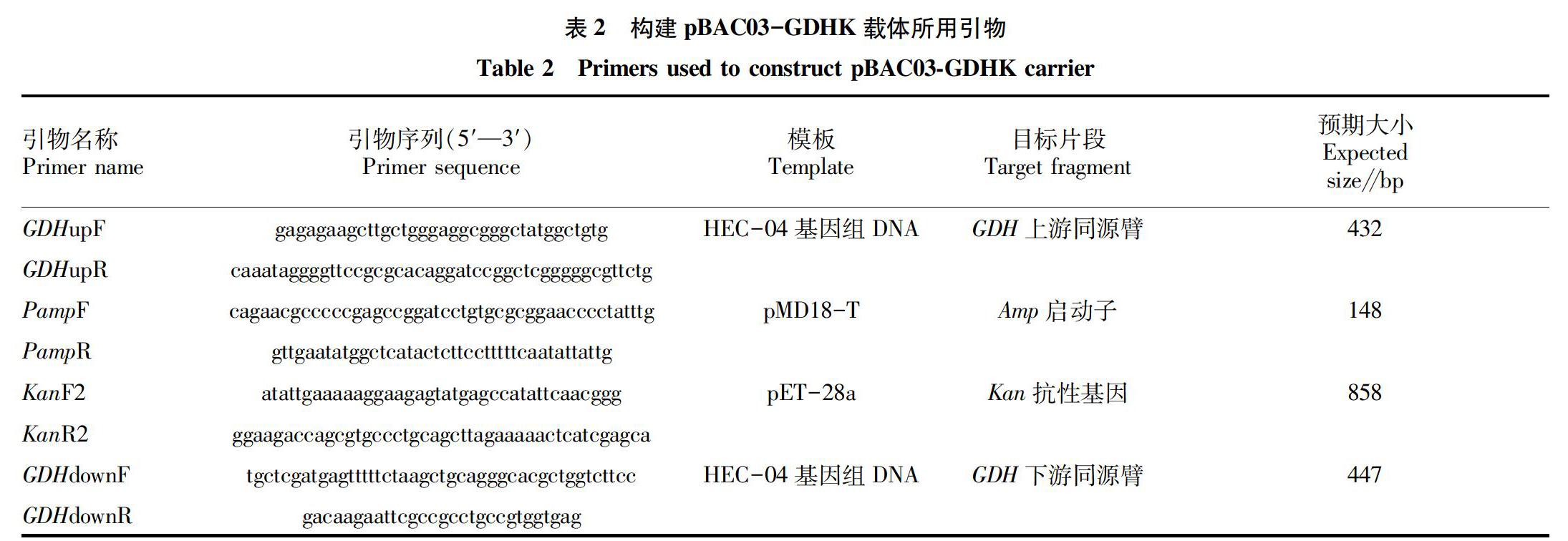
1.2.2 GDH基因敲除载体及敲除片段的构建。
研究参照木醋杆菌基因敲除方案进行[13,16]。通过构建同源重组双交换质粒,用于HEC-004菌株GDH基因的敲除。其中同源敲除盒的结构为GDH上同源臂-Kan表达盒-GDH下同源臂。其中基因组DNA提取、质粒提取、PCR扩增、DNA酶切、回收、连接均参照试剂盒的说明书进行操作。质粒克隆的宿主为按照《分子克隆手册》CaCl2法制备的大肠杆菌DH5a化学感受态。
质粒的具体构建过程如下:按照表2所列的引物和DNA模板分别扩增4个片段并回收,各取0.5 μL作为混合模板,以GDHupF和GDHdownR为引物进行4片段的overlapping PCR扩增。将上述融合PCR产物割胶回收后,利用HindⅢ和EcoRI限制性内切酶进行双酶切,并连接到同样酶切的pMD18-T载体上,在含有50 mg/L Kan和100 mg/L Amp的LB双抗平板上筛选,得到含有pBAC03-GDHKⅡ质粒的克隆。
研究同时也尝试直接采用线性化片段进行基因敲除。将pBAC03-GDHKⅡ质粒进行HindⅢ和EcoRI双酶切,获取包含GDH上游同源臂、Kan抗性表达盒以及GDH下游同源臂的DNA作为线性基因敲除盒(命名GDHKⅡ)。
1.2.3 纳塔葡糖酸醋杆菌感受态的制备及转化。以往文献关于木醋杆菌的转化均用HS培养基(表1)进行感受态细胞制备,后采用电转化的方式进行转化[17-18]。由于研究所使用的菌株为非标准菌株,其感受态细胞的制备及遗传转化的方式需进行条件摸索。一般用处于对数生长期生长活力旺盛的细胞制备电转化感受态细胞。为了筛选出最优的培养基及细胞生长状态,对细胞在3种培养基中的生长状况进行生长曲线考察。
生长曲线的制作方法:从7#平板上挑2个在28 ℃培养了5 d的单克隆,分别转接到含25 mL HS、C2和7#培养基的三角瓶中,同时加入1%纤维素酶溶液防止细菌纤维素的形成,150 r/min,28 ℃培养36 h,然后分别转接10%培养物到新的培养基中继续培养,测定OD600至衰亡期。每种培养基进行3个平行试验。
纳塔葡糖酸醋杆菌HEC-004菌株电转化感受态的制备按照以下方法进行:①将细胞养至对数生长期,用于感受态细胞的制备。②将细胞培养液转入50 mL灭菌离心管中,在預冷的离心机中4 000 r/min离心5 min收集细胞。
③去上清后,用预冷的1 mmol/L HEPES溶液(pH7.0)对细胞轻轻悬浮洗涤,4 ℃ 4 000 r/min离心5 min收集细胞。并将步骤③重复一遍。
④用预冷的15%甘油轻轻重悬细胞,4 ℃ 4 000 r/min离心5 min收集细胞。⑤往收集的细胞沉淀中加入1 mL预冷的15%甘油,置于冰上用于电转化。
⑥在一个预冷的1.5 mL离心管中加入1 μg DNA量的待转化pBAC03-GDHKⅡ质粒或者Hind Ⅲ和Eco RI双酶切的线性化DNA片段,然后加入40 μL做好的感受态细胞,轻轻混匀,转移到预冷的2 mm电转杯中。⑦转化使用Bio-Rad MicroPulser基因导入仪,尝试使用电压:1.5、2.0、2.5和3.0 kV。⑧电击完毕后在电转杯中迅速加入预冷的7#培养基1 mL,将转化后的细胞转移到 PA瓶中进行6 h的复苏培养。⑨6 h后收集菌液,离心浓缩到100 μL,涂布到含有50 mg/L Kan的平板上。
1.2.4 转化子的筛选鉴定。
根据细胞内同源重组的原理,由于pBAC03-GDHKⅡ载体上具有与GDH基因上下游相同的同源臂,因此转化所得到的转化子理论上会出现3种结局:①上游同源臂或者下游同源臂发生了单交换,整个质粒插入到GDH位置(图1A),形成2个拷贝的GDH基因,同时具有Kan和Amp抗性;②按照预期进行双交换;Kan基因替换原GDH基因(图1B),克隆只具有Kan抗性,没有Amp抗性;③发生了随机整合,原GDH基因处未发生变化(图1C),菌株可能同时具有Kan和Amp抗性。而GDHKⅡ线型片段转化子只有2种可能:①按照预期进行了双交换;②发生了随机整合。
由于单交换菌株发生重组后存在正向重复,能发生同源重组,形成回复突变,因此从稳定性方面考虑,目的是筛选进行了双交换的克隆。菌株在含有50 mg/L Kan的抗性平板上生长5 d后,进行克隆的筛选。
采用pBAC03-GDHKⅡ质粒转化得到的转化子,挑取分别接种到含有200 mg/L Amp抗性的C2琼脂平板和50 mg/L Kan抗性的C2琼脂平板进行抗性测试;将只能在Kan平板上生长的转化子用于进一步的PCR基因型验证。线性化片段转化得到的转化子直接采用PCR的方法进行验证。在GDH被替换区域两侧设计引物GDH-F1(5′-cggactgtatcacggtaatcagg-3′)和GDH-R2(5′-caggcggtcctgccacagcac-3′)用于进行整合菌株双交换转化子的鉴定。
1.2.5 GDH基因敲除菌株与出发菌株的产纤维素比较。将筛选到的GDH基因敲除菌株与野生HEC-004菌株进行产纤维素比较试验。在三角瓶中考察纤维素膜的产量,并测定发酵过程的pH,以确定GDH的敲除是否能降低发酵过程葡萄糖酸的产生而改善菌株的产膜能力。
为进一步确定GDH敲除对葡萄糖消耗及葡萄糖酸生成产生的影响,取发酵液200 μL与-20 ℃预冷的丙酮800 μL混匀,-20 ℃冷冻4 h后,12 000 r/min离心10 min进行HPLC检测,分析残液中葡萄糖酸及葡萄糖含量。分析条件:流动相为85%乙腈,色谱柱为Agilent Glycan (4.6×150 mm),柱温为45 ℃,流速为0.5 mL/min, RID检测器温度为35 ℃。
2 结果与分析
2.1 纳塔葡糖酸醋杆菌HEC-004对Kan和Amp的耐受性测定 通过比较发现其在不同抗性浓度下的生长有所不同(表3)。该菌在较高浓度的Amp抗生素培养基中仍然可以生长,在150 mg/L的Amp抗性培养基中培养7 d可以在平板上观察到微菌落。而在Kan抗性条件下,25 mg/L的浓度即可完全抑制菌体生长。综合菌株在2种抗生素平板上的生长情况,在转化子筛选过程中,Kan抗生素的浓度采用25 mg/L,Amp抗生素的浓度采用200 mg/L。
2.2 感受态制备培养基的优化 将HEC-004菌株在HS培养基、7#培养基和C2培养基中进行培养,发现该菌株在7#培养基和C2培养基中可以生长,但在HS培养基中不能生长。为了选择最佳培养基并在合适时间收集细胞制备感受态,绘制了HEC-004在7#和C2培养基的生长曲线(图2)。
从生长曲线上看,7#培养基和C2培养基的最大比生长速率均在μ=0.05。7#培养基的生长曲线显示在35 h后不再生长。综合考虑生长速率和细胞生长状态以及制作感受态细胞所需要的细胞密度,选择C2培养基作为感受态细胞培养用的培养基,当菌体浓度生长到OD600=1.0左右时用于电转化感受态细胞的制备。
2.3 电转化结果
pBAC03-GDHKⅡ质粒图谱见图3。转化产物在含Kan抗生素的7#平板培养5 d后,能看到针尖大小的转化子出现。不同条件下的转化结果如表4所示。pBAC03-GDHKⅡ质粒及其线性化敲除盒在2.0、2.5、3.0 kV电压条件下均获得转化子,其中采用2.5 kV条件下得到的转化子最多。
2.4 阳性克隆的筛选鉴定
通过抗性平板对pBAC03-GDHKⅡ载体转化得到的转化子进行抗性表型筛选。挑选50个克隆在Amp平板上进行点菌,其中有46个具有Amp抗性,剩余的4个采用引物GDH-F1和GDH-R2进行PCR验证,只有3个发生了预期的双交换,双交换概率6%。
GDHKⅡ线型片段所得到的转化子,挑选了40个采用引物GDH-F1和GDH-R2进行PCR验证,结果只有7个克隆具有预期基因型,阳性率为17.5%。
选取其中一个具有正确基因型的阳性克隆HEC-33进行基因组的抽提,采用3组引物进行再鉴定,结果表明HEC-33在3对引物的扩增下均具有预期大小的片段(图4),进一步证明了所挑选克隆为双交换的转化子。该克隆用于下一步的产膜试验验证。
2.5 GDH敲除菌株产纤维素发酵 选取成功进行了双交换的一株菌株HEC-33進行产膜发酵试验。三角瓶中细菌纤维素膜的结果表明,与野生菌株HEC-004相比,HEC-33菌株产膜速度变慢,但最终的纤维素膜产量提升,该结果与Kuo等[13]敲除G.xylinus菌株中的结果类似。经过7 d的培养,与野生菌株相比,HEC-33菌株的产膜湿重提高了18.3%。通过肉眼观察,工程菌株所产膜质地更均匀,柔韧性更好。对HEC-004和HEC-33菌株在三角瓶中培养的纤维素膜发酵残液进行pH测定,也证明敲除了GDH基因的菌株,其发酵残液中没有葡萄糖酸的产生(表5)。
3 讨论
该研究通过培养基筛选确定了高产细菌纤维素的G.nataicola 菌株HEC-004的感受态制备方法。由于目前暂未查询到有文献研究G.nataicola的遗传转化方法,试验首先借鉴了文献报道常用于培养Gluconacetobacter属的HS培养基[12-13]对HEC-004菌株进行培养,结果表明HEC-004菌株在HS培养基中难以生长,说明HEC-004菌株与目前文献报道的Gluconacetobacter属其他菌株在代谢生理上存在区别。
不少研究中从自然界中分离产纤维素微生物时采用的均为HS培养基[19],而该研究中的高产纤维素菌株在该类型中不能生长,这可能导致高产菌株的漏筛,因此该研究结果可为后续菌株筛选提供借鉴。
以往的研究文献中,产细菌纤维素菌株的基因敲除都是采用带同源臂的质粒进行转化[13,16]。该研究尝试线性化片段进行直接电转化,也可以得到阳性转化子,证明了可以通过直接转线性化片段对G.nataicola进行基因敲除或者敲入。由于线性化片段不会出现单交换克隆子,因此在该研究中,转化子中采用线性片段进行转化的双交换概率大于质粒转化试验组(17% vs 6%)。该结果也说明,G.nataicola菌的同源重组概率较低,相对而言,大多数转化子均发生了非同源重组插入到了基因组的未知位置。在后期研究中,可借助目前广泛使用的基因编辑方法,在该类菌株中建立基因编辑系统,提高对菌株定向操作的能力。
GDH基因负责编码G.nataicola菌株中的葡萄糖脫氢酶,使发酵培养基中的葡萄糖变为葡萄糖酸。试验中敲除GDH后所得到的HEC-33菌株生长速度变慢,产膜速度也变慢,这可能与GDH基因编码的葡萄糖脱氢酶在葡萄糖脱氢过程中产生还原型PQQ辅酶能促进细胞生长有关[20],敲除该基因后影响了还原型PQQ的生成,因此菌体生长速度变慢。但是发酵结束时,工程菌株的膜产量比野生型高,推测野生型菌株中葡萄糖酸的产生影响了发酵体系的pH,使得体系过酸,导致后期菌体活力下降,产膜能力受影响。
4 结论
以一株工业化高产细菌纤维素的纳塔葡糖酸醋杆菌为对象,研究了其遗传转化条件,并敲除了其中与葡萄糖酸生成相关的葡萄糖脱氢酶基因GDH,构建了一株在长周期发酵过程中具有更高产膜能力的工程菌株。
参考文献
[1] ROSS P,MAYER R,BENZIMAN M.Cellulose biosynthesis and function in bacteria[J].Microbiol reviews,1991,55(1):35-58.
[2] CZAJA W,KRYSTYNOWICZ A,BIELECKI S,et al.Microbial cellulose-the natural power to heal wounds[J].Biomaterials,2006,27(2):145-151.
[3] RAJWADE J M,PAKNIKAR K M,KUMBHAR J V.Applications of bacterial cellulose and its composites in biomedicine[J].Applied microbiology and biotechnology,2015,99(6):2491-2511.
[4] KUCIN'SKALIPKA J,GUBANSKA I,JANIK H.Bacterial cellulose in the field of wound healing and regenerative medicine of skin:Recent trends and future prospectives[J].Polymer bulletin,2015,72(9):2399-2419.
[5] FU L N,ZHANG Y,LI C,et al.Skin tissue repair materials from bacterial cellulose by a multilayer fermentation method[J].Journal of materials chemistry,2012,22(24):12349-12357.
[6] CLEENWERCK I,DE VOS P,DE VUYST L.Phylogeny and differentiation of species of the genus Gluconacetobacter and related taxa based on multilocus sequence analyses of housekeeping genes and reclassification of Acetobacter xylinus subsp.sucrofermentans as Gluconacetobacter sucrofermentans (Toyosaki et al.1996) sp.nov.,comb.nov.[J].International journal of systematic and evolutionary microbiology,2009,60:2277-2283.
[7] KWAK M H,KIM J E,GO J,et al.Bacterial cellulose membrane produced by Acetobacter sp.A10 for burn wound dressing applications[J].Carbohydrate polymers,2015,122:387-398.
[8] 向东,王锡彬,钟春燕.不同供氧方式对木醋杆菌培养中生物量的影响[J].现代食品科技,2011,27(8):873-876.
[9] 吴敏.水溶性多糖对生物合成细菌纤维素的影响[D].海口:海南大学,2011.
[10] 牛成,吴周新,王锡彬,等.木醋杆菌菌株的特性研究[J].海南师范大学学报(自然科学版),2009,22(1):55-59.
[11] 王志国,韦燕来,韦茂山,等.有机酸在合成培养基中对木醋杆菌合成纤维素的影响[J].食品科学,2008,29(5):295-297.
[12] CHIEN L J,CHEN H T,YANG P F,et al.Enhancement of cellulose pellicle production by constitutively expressing Vitreoscilla hemoglobin in Acetobacter xylinum[J].Biotechnology progress,2006,22(6):1598-1603.
[13] KUO C H,TENG H Y,LEE C K.Knockout of glucose dehydrogenase gene in Gluconacetobacter xylinus for bacterial cellulose production enhancement[J].Biotechnology and bioprocess engineering,2015,20(1):18-25.
[14] FLOREA M,HAGEMANN H,SANTOSA G,et al.Engineering control of bacterial cellulose production using a genetic toolkit and a new celluloseproducing strain[J].Proceedings of the national academy of sciences,2016,113(24):3431-3440.
[15] SCHRAMM M,HESTRIN S.Factors affecting production of cellulose at the air/liquid interface of a culture of Acetobacter xylinum[J].Journal of general microbiology,1954,11(1):123-129.
[16] SHIGEMATSU T,TAKAMINE K,KITAZATO M,et al.Cellulose production from glucose using a glucose dehydrogenase gene (gdh)deficient mutant of Gluconacetobacter xylinus and its use for bioconversion of sweet potato pulp[J].Journal of bioscience and bioengineering,2005,99(4):415-422.
[17] CASTRO C,CLEENWERCK I,TRCEK J,et al.Gluconacetobacter medellinensis sp nov.,cellulose and noncelluloseproducing acetic acid bacteria isolated from vinegar[J].International journal of systematic and evolutionary microbiology,2013,63(Pt 3):1119-1125.
[18] SUNAGAWA N,FUJIWARA T,YODA T,et al.Cellulose complementing factor (Ccp) is a new member of the cellulose synthase complex (terminal complex) in Acetobacter xylinum[J].Journal of bioscience and bioengineering,2013,115(6):607-612.
[19] AYDIN Y A,AKSOY N D.Utilization of vinegar for isolation of cellulose producing acetic acid bacteria[J].AIP Conference Proceedings,2010,1247(1):340-348.
[20] DUINE J A,VAN DER MEER R A,GROEN B W.The cofactor pyrroloquinoline quinone[J].Annual review of nutrition,1990,10:297-318.
