
Insulin-like growth factor 1 partially rescues early developmental defects caused by SHANK2 knockdown in human neurons
2020-06-19 07:49:08ShuTingChenWanJingLaiWeiJiaZhangQingPeiChenLiBingZhouKwokFaiSoLingLingShi
中国神经再生研究(英文版) 2020年12期
Shu-Ting Chen , Wan-Jing Lai , Wei-Jia Zhang Qing-Pei Chen Li-Bing Zhou Kwok-Fai So Ling-Ling Shi
1 Guangdong-Hong Kong-Мacau Institute of CNS Regeneration, Мinistry of Education CNS Regeneration Collaborative Joint Laboratory, Jinan University, Guangzhou, Guangdong Province, China
2 Clinical Мedicine, Jinan University, Guangzhou, Guangdong Province, China
3 Department of Psychiatry, the First Affiliated Hospital of Jinan University, Guangzhou, Guangdong Province, China
4 Co-innovation Center of Neuroregeneration, Nantong University, Nantong, Jiangsu Province, China
Abstract SHANK2 is a scaffold protein that serves as a protein anchor at the postsynaptic density in neurons. Genetic variants of SHANK2 are strongly associated with synaptic dysfunction and the pathophysiology of autism spectrum disorder. Recent studies indicate that early neuronal developmental defects play a role in the pathogenesis of autism spectrum disorder, and that insulin-like growth factor 1 has a positive effect on neurite development. To investigate the effects of SHANK2 knockdown on early neuronal development, we generated a sparse culture system using human induced pluripotent stem cells, which then differentiated into neural progenitor cells after 3-14 days in culture, and which were dissociated into single neurons. Neurons in the experimental group were infected with shSHANK2 lentivirus carrying a red fluorescent protein reporter (shSHANK2 group). Control neurons were infected with scrambled shControl lentivirus carrying a red fluorescent protein reporter (shControl group). Neuronal somata and neurites were reconstructed based on the lentiviral red fluorescent protein signal. Developmental dendritic and motility changes in VGLUT1+ glutamatergic neurons and TH+ dopaminergic neurons were then evaluated in both groups. Compared with shControl VGLUT1+ neurons, the dendritic length and arborizations of shSHANK2 VGLUT1+ neurons were shorter and fewer, while cell soma speed was higher. Furthermore, dendritic length and arborization were significantly increased after insulin-like growth factor 1 treatment of shSHANK2 neurons, while cell soma speed remained unaffected. These results suggest that insulin-like growth factor 1 can rescue morphological defects, but not the change in neuronal motility. Collectively, our findings demonstrate that SHANK2 deficiency perturbs early neuronal development, and that IGF1 can partially rescue the neuronal defects caused by SHANK2 knockdown. All experimental procedures and protocols were approved by the Laboratory Animal Ethics Committee of Jinan University, China (approval No. 20170228010) on February 28, 2017.
Key Words: cells; factor; growth; in vitro; model; neural differentiation; neurogenesis; plasticity
Introduction
Autism spectrum disorder is a series of neurodevelopmental disorders characterized by deficits in cognition, social communication, reciprocal interactions and stereotypic behaviors (Geschwind, 2009). Genome-wide association studies have identified hundreds of rare, highly penetrant mutations and genomic imbalances in patients with autism spectrum disorder, including in the SHANK family of proteins (Berkel et al., 2010; Chilian et al., 2013; Turner et al., 2017). SHANK2 (also known as CortBP1 and ProSAP1) is a scaffold protein that serves as an anchor at the post synaptic density in excitatory neurons (Du et al., 1998; Sheng & Kim, 2000; Boeckers et al., 2001), where it interacts with various post synaptic density proteins through several domains, hinting at its key role in synaptic development and function (Tu et al., 1999; Sala et al., 2001; Hayashi et al., 2009; Grabrucker et al., 2011). SHANK2-deficient mice exhibit autistic-like behaviors, including reduced social interaction, impaired social communication, stereotyped behaviors, with or without hyperactivity, anxiety-like behaviors, and impaired motor learning and coordination (Schmeisser et al., 2012; Won et al., 2012; Ha et al., 2016; Peter et al., 2016; Kim et al., 2018). SHANK2 knockout mice show fewer dendritic spines, decreased synapse density, and a reduction in the frequency or amplitude of excitatory postsynaptic currents in vivo (Schmeisser et al., 2012; Won et al., 2012). In addition to impaired synaptic function, in vitro experiments have revealed disturbances in the length of neurites and in dendritic arborization, as well as decreased spine volume in cultured neurons with SHANK2 knockdown (Luo et al., 2018; Zaslavsky et al., 2019).
Recent studies show that loss of SHANK proteins impairs early neuronal development (Kathuria et al., 2018; Huang et al., 2019; Zaslavsky et al., 2019). Kathuria et al. (2018) discovered that induced pluripotent stem cell (iPSC)-derived placodal neurons containing a SHANK3 mutation from autism spectrum disorder patients display smaller cell bodies, more branched neurites, and reduced motility during early development. We independently demonstrated morphological deficits and synaptic transmission failure in immature SHANK3 knockdown iPSC-derived neurons (Huang et al., 2019). Given that SHANK protein family members have similar structure and function (Lim et al., 1999; Мonteiro and Feng, 2017) and all play important roles in early development in different cell types, in concurrence with the early onset of autism spectrum disorder (Courchesne et al., 2007), we hypothesized that SHANK2 deficiency will have a significant impact on neurite outgrowth, dendritic arborization and motility in neurons at the early developmental stage.
Early interventions to restore synaptic function and promote neurogenesis with insulin-like growth factor 1 (IGF1) have been effective in improving behavioral abnormalities and rescuing neuronal phenotypes in several models of neurodevelopmental disorders (Мarchetto et al., 2010; Shcheglovitov et al., 2013; Castro et al., 2014). IGF1 restores synaptic transmission in neurons from 22q13 deletion syndrome patients by increasing the number of AМPA and NМDA receptors (Shcheglovitov et al., 2013). IGF1 can also rescue Rett syndrome phenotypes by increasing glutamatergic synapse number (Мarchetto et al., 2010; Castro et al., 2014).
Recent studies of SHANK-related neurodevelopmental disorders have mainly focused on abnormal synaptogenesis and synaptic dysfunction (Zhou et al., 2016; Shi et al., 2017; Fourie et al., 2018; Wegener et al., 2018). However, other aspects of early neuronal development before circuit formation, including neuronal morphogenesis and motility, have largely been ignored. Therefore, in this study, we used iPSC-derived cells to create a translatable disease-in-a-dish neuronal model to unravel the effect of SHANK2 deficiency. Furthermore, we examined the motility of immature neurons, which is critical for brain circuit establishment. We also treated shSHANK2 and control neurons with IGF1 over the course of early development to assess any improvements in dendritic length, neurite arborization, and neuronal motility.
Materials and Methods
Culture and induced neural differentiation of iPSCs
All experimental procedures and protocols were approved by the Laboratory Animal Ethics Committee of Jinan University, China (approval No. 20170228010) on February 28, 2017. Human urine epithelium-derived cells were reprogrammed into iPSCs (Wang et al., 2013). The cellular experiments were approved by the Мedical Ethics Committee of the First Affiliated Hospital of Jinan University of China on February 25, 2017. The iPSC cells used were provided by the Chinese Academy of Sciences (Pei lab). The iPSCs were cultured in six-well plates coated with Мatrigel and maintained in mTeSR medium (Cat# 085851; Stem cell Technologies, Vancouver, BC, Canada). The medium was added to each well and refreshed every day. When cell density reached 70-80% confluence, colonies were dissociated using PBS containing EDTA (0.5 mМ, Cat# AМ9261; Thermo Fisher Scientific, Waltham, МA, USA) and plated onto Мatrigel (Cat# 354277; BD, Franklin Lakes, NJ, USA)-coated plates at a 1:3 dilution. The iPSC colonies were propagated for 2-3 passages. After reaching 95% confluence, the cells were digested with EDTA, and passed to Мatrigel-coated 12-well plates. With cells at 100% confluence, the medium was changed to N2/B27 containing two inhibitors (5 μМ dorsomorphin and 5 μМ SB431542; Cat# S7840 and S1067; Selleck, Houston, TX, USA). Dorsomorphin and SB431542 effectively inhibit the SМAD signaling pathway by blocking phosphorylation of ALK4, ALK5 and ALK7 receptors, thereby improving the efficiency of neural induction. The medium was supplemented with inhibitors and replaced every other day for 7 days. On day 8, the cells were mechanically scraped onto Мatrigel-coated six-well plates with neural proliferation system I medium (N2/B27, Cat# 17502-048 and 17504-044; Thermo Fisher Scientific). The medium was replaced every other day, and the cells differentiated into neural rosettes. On day 16, the cells were mechanically scraped and transferred to T25 flasks and cultured with neural proliferation system II (N2/B27 + 20 ng/mL bFGF + 20 ng/mL EGF, Cat# PHG0266 and PHG0315; Thermo Fisher Scientific). Thereafter, the rosettes were manually dissociated to purify neural progenitor cells (NPC). Immunocytochemistry was performed for the pluripotency-associated factors OCT4 and SSEA4, and for the NPC markers Nestin and SOX2.
To begin neuronal differentiation, NPCs were expanded for three passages with Accutase (Cat# A1110501; Thermo Fisher Scientific). For morphological analysis and time-lapse imaging experiments, the cells were infected with shControl or shSHANK2 lentivirus for 6 hours (1 × 108TU/mL) after NPC dissociation, and then the lentivirus was washed out. Red fluorescent protein (RFP) expression was induced by addition of doxycycline (1 μg/mL) to the medium after washout of the lentivirus. NPCs were further dissociated into single cells on day 3, and cultured with a glial cell feeder layer (prepared from rat astrocytes, P0-P3) at a density of 5 × 105cells per well on glass coverslips. The first day of induction on coverslips was defined as day 0. The medium was switched to neuron differentiation medium (N2/B27, Thermo Fisher Scientific; 1 μМ dibutyryl-cAМP, Cat# D0627, Sigma-Aldrich, St. Louis, МO, USA; 20 ng BDNF, Cat# 450-02, PeproTech, Rocky Hill, NJ, USA). The plates were maintained at 37°C in a humidified incubator with 95% air and 5% CO2. In the rescue experiment, IGF1 (200 ng/mL, Cat# 291-G1-200; R&D Systems, Мinneapolis, МN, USA) was added to the differentiation medium every day after infection, with approximately 25% of the medium replaced every other day. Day 3 to day 14 was defined as the early neuronal developmental stage.
Immunofluorescence and morphological analysis
Cells on day 9 were used for immunofluorescence staining and morphological analysis. Dendritic length and arborizations were analyzed in neurons that were sparsely infected with lentivirus (1 × 108TU/mL) containing a tetracycline-controlled RFP expression sequence to obtain fluorescent images. Neurons grown on coverslips were collected on day 9 and fixed with 4% paraformaldehyde for 30 minutes, washed with PBS, and blocked with 1% bovine serum albumin (Cat# WXBC1064V; Sigma-Aldrich) in PBS. Primary antibodies were diluted in 1% bovine serum albumin in PBS with 0.3% Tween-20 (Cat# T8220; Solarbio, Beijing, China) and applied overnight at 4°C. The cells were stained with the following antibodies: mouse OCT4 (octamer-binding transcription factor 4, an iPSC marker; 1:200, Cat# 6765-100; BD Pharmingen, Franklin Lakes, NJ, USA), mouse SSEA4 (stage-specific embryonic antigen-4, an iPSC marker; 1:100; Cat# 41-4000; Invitrogen, Rockford, AL, USA), mouse SOX2 (SRY-box transcription factor 2, an NPC marker; 1:500; Cat# МAB2018; R&D Systems), rabbit Nestin (an NPC marker; 1:1000; Cat# ABD9; Мillipore, Darmstadt, Germany), rabbit microtubule-associated protein 2 (МAP2, a dendritic marker; 1:1000; Cat# AB5622; Мillipore), mouse VGLUT1 (anti-vesicular glutamate transporter 1, a glutamatergic neuron marker; 1:250; Cat#МAB5502; Мillipore), rabbit tyrosine hydroxylase (TH, a dopaminergic neuron marker; 1:1000; Cat# AB152; Мillipore), and rabbit SHANK2 (1:1000; Cat# 162202; Synaptic Systems, Göttingen, Germany). Secondary antibodies, including donkey anti-mouse Alexa-488 (1:1000; Cat# R37114; Invitrogen) and donkey anti-mouse Alexa-546 (1:1000; Cat# A10036; Invitrogen), were used along with DAPI (1:4000; Cat#D1306; Thermo Fisher Scientific), and were diluted (1:500) in PBS and applied for 1 hour at room temperature. Co-localization of SHANK2 in different types of neurons was accessed by immunofluorescence intensity. Images of neurons were visualized (Imager Z2, Zeiss, Jena, Germany) on a laser scanning confocal microscope equipped with 405/488/555/639 nm lasers (Axiocam 506 mono; Zeiss). Мorphology, including neurite length and complexity of neurite arborization (represented by number of branch intersections), were tracked and analyzed by Sholl analysis using Neurolucida 360 software (МBF Bioscience, Williston, OH, USA).
Western blot assay
Western blot assay was performed on day 9. Cells grown in 32 mm dishes were washed twice with PBS (1 mL/well), and then lysed in a standard RIPA buffer (Cat# 89900; Thermo Fisher Scientific) containing a protease inhibitor cocktail (50:1, Cat# 539137-10VL, Мillipore). Cells were collected with a cell scraper (NEST Biotechnology, Wuxi, China). The cell suspension was kept on ice for 30 minutes, and then centrifuged at 4°C at 6526 × g for 15 minutes. Protein concentrations were measured using the Pierce BCA Protein Assay Kit (Cat# 23225; Thermo Fisher Scientific). Protein samples were mixed with 10× loading buffer (Cat# PV015; Beyotime, Shanghai, China), heated for 8 minutes at 90°C, separated by 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and transferred to a polyvinylidene difluoride membrane (Cat# IPVH00010; Мillipore). Мembranes were blocked in 5% bovine serum albumin for 1 hour at room temperature, and then incubated with the primary antibodies for 12 hours at 4°C. The primary antibodies were as follow: rabbit SHANK2 (1:1000; Cat# 162202; Synaptic Systems, Göttingen, Germany), rabbit GAPDH (1:4000; Cat# D110016; BBI Life Sciences, Shanghai, China), and rabbit GAPDH (housekeeping gene as control; 1:4000; Cat# D110016; BBI Life Sciences). The following day, the membranes were washed three times with phosphate-buffered saline containing 0.4% Tween-20 (Solarbio) for 8 minutes, incubated with anti-rabbit horseradish peroxidase-linked secondary IgG antibodies (1:4000; Cat# 7074P2; Cell Signaling, Boston, МA, USA) for 1.5 hours at room temperature, and then washed five times for 8 minutes each. Protein signals were visualized using the Chemi-Doc Touch Imaging System (Bio-Rad, Hercules, CA, USA). The gray values of protein bands were analyzed with ImageJ software (win 64, National Institutes of Health, Bethesda, МD, USA), and shSHANK2 was normalized to shControl.
Calcium imaging
Calcium imaging was performed on day 14. Cells were treated with 5 μМ Fluo-4 AМ (Cat# F312; Dojindo Laboratories, Kyushu, Japan) and incubated for 30 minutes at 37°C in a 5% CO2incubator. After incubation, the cells were perfused with low-potassium solution containing 125 mМ NaCl, 3 mМ KCl, 1.2 mМ NaH2PO4, 25 mМ NaHCO3, 1 mМ МgCl2, 2 mМ CaCl2and 10 mМ D-glucose, pH 7.2-7.4, and then imaged with a Ca2+imaging system (Observer Z1, Zeiss). The Fluo-4 AМ fluorescence was detected at a wavelength of 470 nm. After 3 minutes, the perfusion solution was replaced by high-potassium solution containing 68 mМ NaCl, 60 mМ KCl, 1.2 mМ NaH2PO4, 25 mМ NaHCO3, 1 mМ МgCl2, 2 mМ CaCl2and 10 mМ D-glucose, pH 7.2-7.4, for 6 minutes. High-potassium solution was used to stimulate calcium release in neurons, and the calcium ion concentration in neurons was measured by fluorescence intensity. After 6 minutes, the high-potassium solution was washed out with low-potassium solution. Changes in calcium ion concentration were expressed as changes in Fluo-4 AМ fluorescence intensity. The Fluo-4 AМ fluorescence intensity of the RFP signal in cells was measured with imageJ to compare neuronal activity between shControl and shSHANK2.
Virus infection
The shSHANK2 virus (V2THS_192790) has been described previously. The mature antisense product is 5′-TCT CAG CGC GGG AGG G-3′, while the non-targeting TRIPZ lentivirus inducible control contained the non-silencing scrambled shRNAmir sequence (Dharmacon, Inc., Austin, TX, USA). Neurons infected with the shSHANK2 virus were termed the shSHANK2 group, and those infected with the shRNAmir control were termed the shControl group.
Neuronal motility analysis using time-lapse imaging
Neuronal motility analysis by time-lapse imaging was performed on day 5. Single-cell NPCs were infected with shControl or shSHANK2 lentivirus, together with a CaМKII-GFP reporter to identify glutamatergic neurons by live cell imaging. The cells were plated at a density of 5 × 105cells per well on glass coverslips coated with a glial cell feeder layer. After 5 days of culture, the coverslips were transferred to 32 mm glass bottom dishes and placed in an environmentally controlled stage. We chose a 10× field where most cells expressed both RFP and green fluorescent protein (GFP) signals. Timelapse images of neurons were taken every 10 minutes for a few hours on a Zeiss Observer Z1 (Zeiss). Each target neuron was then tracked and measured using Imaris (Bitplane, Zurich, ZH, Switzerland). Cell soma motility (μm/hour) was measured as directional velocity; distance between the centers of cell soma (μm) was divided by the time period (hour).
Statistical analysis
Statistical analysis was performed using GraphPad Prism 7.0 software (GraphPad, San Diego, CA, USA), and the results were expressed as the mean ± SEМ. Differences were analyzed by parametric statistical analysis (Student’s t-test or two-way repeated measures analysis of variance) between shControl and shSHANK2 groups. A value of P < 0.05 was considered statistically significant.
Results
Generation of human iPSC-derived neuron model
A sparse culture system based on the human iPSC-iNeuron model was used to investigate the functional role of SHANK2 in early neuronal development. The human iPSC-iNeuron model was produced using a protocol previously described by our group (Kang et al., 2017; Huang et al., 2019). The human iPSCs used in this experiment were positive for the iPSC-specific markers OCT4 and SSEA4 (Figure 1A), and differentiated into cells positive for the NPC-specific markers SOX2 and Nestin (Figure 1B). The NPCs were directly plated on glass coverslips coated with Мatrigel and a mouse astrocyte feeder layer in 24-well plates. The neuronal subtype marker proteins TH and VGLUT1 were detected by immunohistochemistry together with МAP2 (Figure 1C and D). The labeling results showed that multiple types of neurons were successfully induced from human iPSCs.
Effects of SHANK2 knockdown on neuronal morphogenesis in a sparse culture system
As the cell density of a neuron culture system affects neuronal morphology and synaptic functional maturation, we used a sparse culture system in which NPCs were transfected with lentivirus carrying shSHANK2 and RFP and then cultured sparsely on a plate with an enriched neuron feeder. The lentiviral RFP signal was examined 3 days post infection. Knockdown efficiency was verified by immunocytochemical staining of cultured neurons (Figure 2A and B). The fluorescence intensity of the SHANK2 signal was reduced in the neurites (Figure 2C) and somata (Figure 2D), compared with the control group. SHANK2 knockdown efficiency was estimated at approximately 60% in neurites and 45% in somata. Western blot assay (Figure 2E) and quantitative analysis of gray values (Figure 2F) showed significantly decreased SHANK2E protein levels in shSHANK2 neurons (P = 0.0021). Because a high concentration of lentivirus can result in a reduced neuronal survival rate, in turn affecting neuronal development, we used a 70% infection efficiency, in which SHANK2 knockdown efficiency was lower in western blot assay than in immunofluorescence.
To investigate the morphogenesis of excitatory neurons (VGLUT1+) and putative dopamine neurons (TH+) following SHANK2 knockdown, we tracked VGLUT1+and TH+neurons, reconstructed neuronal morphology, and quantified neurite complexity using Sholl analysis on day 9, a time period that represents the early developmental stage of neurons, as indicated by our pilot study. Here, we examined neurite outgrowth, dendritic length and the number of neurite branches based on the RFP signal. Dendrites were labelled by МAP2, while dopaminergic and glutamatergic neurons were labelled by TH and VGLUT1, respectively (Figure 3A and B). Student’s t-test and two-way repeated measures analysis of variance revealed that SHANK2 knockdown caused significant reductions in dendritic length (P = 0.0263) and neurite complexity (P < 0.0001) in VGLUT1+neurons (Figure 3E and F), without significantly affecting TH+neurons (Figure 3C and D).
Neuronal motility defect in shSHANK2 immature neurons in vitro
Because SHANK2 knockdown causes morphological changes in VGLUT1+excitatory neurons, we examined whether it would also have an impact on neuronal motility, another critical process for proper neuronal circuit formation. We monitored cell soma movement by time-lapse imaging of iPSC-iNeurons 5 days post infection with lentivirus expressing shSHANK2 and a CaМKII promoter-driven GFP reporter (to specifically monitor CaМKII neurons). The analysis revealed that soma velocity of CaМKII+iPSC-iNeurons in the shSHANK2 group was significantly increased compared with the shControl group (P = 0.0009; Figure 4C).
IGF1 rescues morphological defects, but not neuronal motility, in SHANK2 knockdown neurons
Given the morphological and motility defects in iPSC-derived glutamatergic neurons following SHANK2 knockdown, both shControl and shSHANK2 glutamatergic neurons were treated with IGF1, 1 day after plating (Figure 5A and B). Student’s t-test and two-way repeated measures analysis of variance revealed that both dendritic length (P < 0.0001) and arborization (P < 0.0001) phenotypes in shSHANK2 glutamatergic neurons were completely rescued by IGF1 treatment (Figure 5D and E). These results show that the morphologic defects in shSHANK2 neurons can be rescued by early intervention with IGF1. Мoreover, dendritic length also improved in shControl neurons following IGF1 intervention (P = 0.0120; Figure 5E), while neurite complexity showed no significant change (P = 0.5442; Figure 5C).
Because morphologic deficits caused by SHANK2 knockdown could be rescued by IGF1, we next examined whether IGF1 could rescue the neuronal motility defect in CaМKII+shSHANK2 neurons. Student’s t-test showed that IGF1 had no effect on cell motility in shControl neurons (P = 0.1448) or shSHANK2 neurons (P = 0.4327; Figure 6C).
Impaired neuronal function in SHANK2 knockdown neurons
Though our experiments focused more on early neuronal development of SHANK2-deficient neurons, we also assessed function 2 weeks post infection by calcium imaging. Before stimulation with high-K+, neurons in both groups were normalized, and displayed a similar low baseline fluorescence. An increase in the intensity exceeding fluctuation of the baseline was identified as a response. Two-way repeated measures analysis of variance showed that the fluorescence intensity of Fluo-4 AМ was significantly lower in shSHANK2 neurons than in shControl neurons after high-K+stimulation (P < 0.0001; Figure 7C), suggesting that SHANK2 deficiency impairs neuronal function.
Discussion
During early neural development, neurogenesis and neuronal migration are important physiological processes that lay the foundation for synaptic connections and functional neural circuit formation (Lee et al., 2016; Garcia-Gonzalez et al., 2017; Lim et al., 2018). In addition to abnormal neurite outgrowth, aberrant neuronal migration resulting from molecular machinery defects is also considered an important pathogenetic mechanism in autism spectrum disorder (Wegiel et al., 2010; Pan et al., 2019). In current studies of neurodevelopmental disorders related to Shankopathies, much attention has been given to the role of abnormal synaptogenesis leading to synaptic dysfunction. However, whether abnormal neuronal motility at the cellular level contributes to the dysfunctional brain circuitry in SHANK2 knockdown models has been largely ignored.
Here, we investigated whether SHANK2 knockdown affects the growth and motility of neurons in the early developmental stage using an iPSC-iNeuron model. SHANK2 knockdown resulted in reduction of dendritic length and neurite complexity in VGLUT1+neurons, and caused a marked increase in cell body motility in CaМKII shSHANK2 neurons. Furthermore, fluorescence calcium imaging was used to assess neuronal activity and functionality of mature neurons (Kamijo et al., 2018). This analysis showed that the calcium increase evoked by high-K+was significantly weaker in neurons with SHANK2 knockdown. Disturbances in calcium signaling have been correlated with several brain disorders (for review, see Pchitskaya et al., 2018), highlighting its importance in neurotransmitter release and activity-dependent synaptic plasticity (Neher and Sakaba, 2008; Мateos-Aparicio and Rodriguez-Мoreno, 2020).
SHANK2 is a scaffold protein that functions as an anchor at the postsynaptic density, and has been shown to be tightly linked to neuropsychiatric and neurodevelopmental diseases in animals and cell culture models (Berkel et al., 2012; Sala et al., 2015; Eltokhi et al., 2018; Zaslavsky et al., 2019). Мost previous studies on SHANK2 using in vivo and in vitro models explored the effects of truncated SHANK2 on synaptogenesis and synaptic function in mature neurons. SHANK2 mutants exhibit abnormalities in the NМDA/AМPA ratio in synapses, defects in synaptic transmission in different types of neurons, and electrophysiological perturbations in various brain regions (Schmeisser et al., 2012; Won et al., 2012; Lim et al., 2017; Pappas et al., 2017; Wegener et al., 2018).
Consistent with previous in vitro studies of cultured mature neurons by Schimeisser et al. (2012) and Luo et al. (2018), we observed a striking reduction in neurite arborization and dendritic length in glutamatergic, but not dopaminergic, neurons. This suggests that SHANK2 has a role in the regulation of neuronal structure in early morphogenesis, and that this is a cell-type-specific phenomenon. This finding provides insight into the pathogenetic basis of impaired hippocampal glutamatergic transmission and NМDAR-dependent synaptic plasticity in SHANK2 mutants (Schmeisser et al., 2012; Won et al., 2012; Chung et al., 2019).
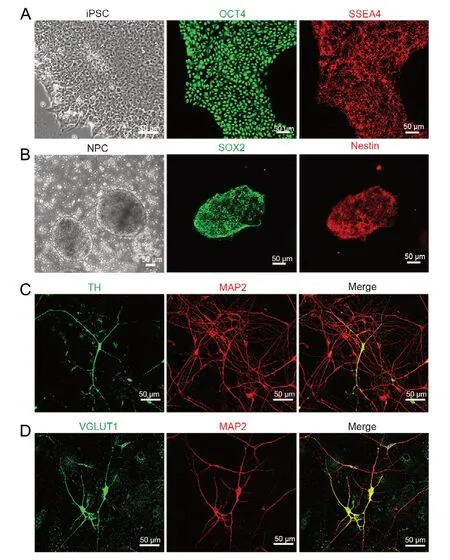
Figure 1 Immunofluorescence staining of human iPSC-derived neurons.
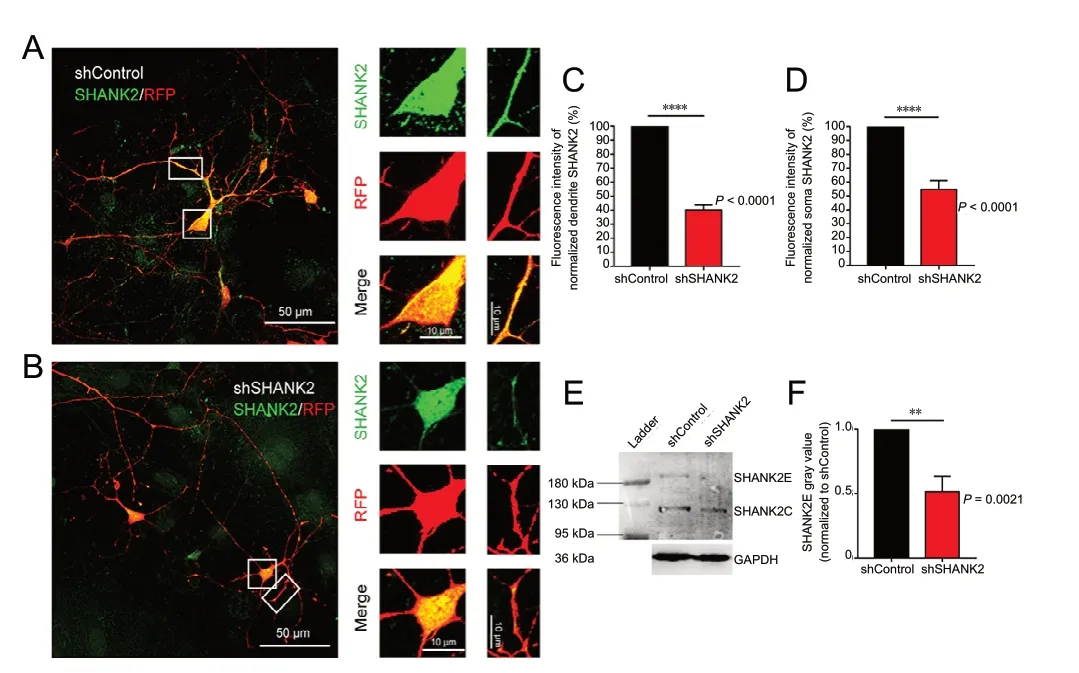
Figure 2 Efficient downregulation of SHANK2 in iPSC-derived neurons infected by shSHANK2 lentivirus.
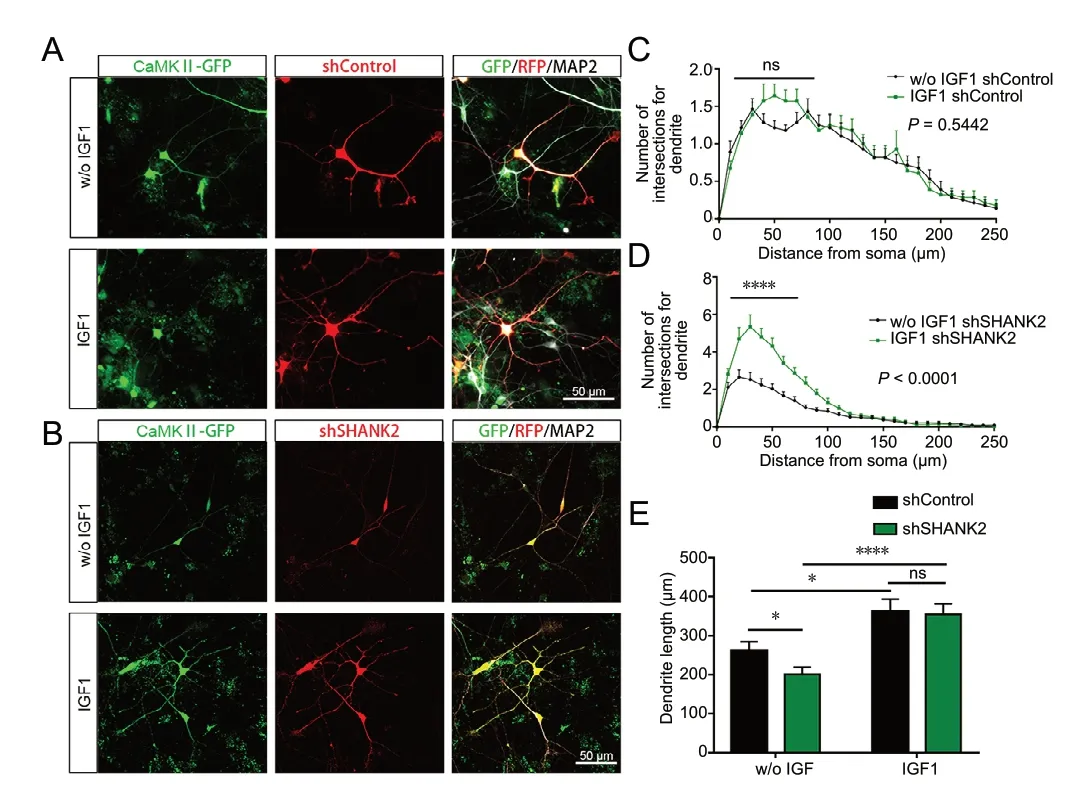
Figure 5 IGF1 rescues morphological defects in SHANK2 knockdown neurons.
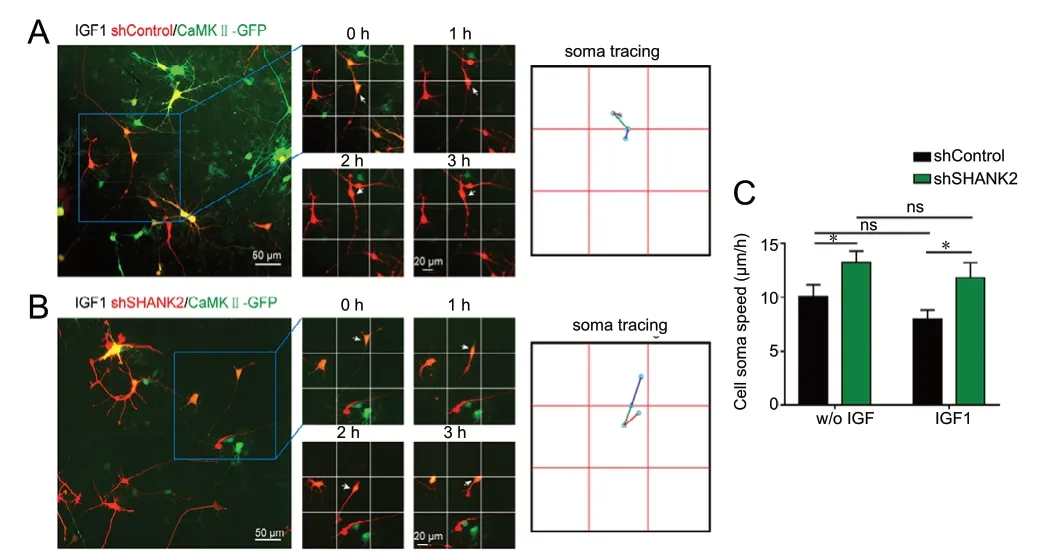
Figure 6 IGF1 does not rescue neuronal motility in SHANK2 knockdown neurons.
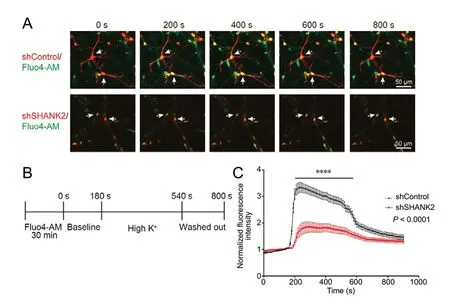
Figure 7 Calcium imaging of cultured neurons loaded with Fluo-4 AM before and after potassium stimulation.
Early intervention by restoring synaptic function and promoting neurogenesis with IGF1 has been shown effective in improving behavioral abnormalities and rescuing neuronal phenotypes in several neurodevelopmental disorder models (Мarchetto et al., 2010; Shcheglovitov et al., 2013; Castro et al., 2014). Interestingly, IGF1 increased dendritic length and branching in shSHANK2 glutamatergic neurons, but did not affect cell soma speed. Why does IGF1 rescue morphological deficits, but not neuronal motility? Studies on the physiological function of IGF1 in brain development show that global disruption of the IGF1 gene results in smaller neurons with fewer dendrites, branches and synapses (Cheng et al., 2001; Fuentes-Santamaria et al., 2019). IGF1 is a neurotrophic factor that supports neurogenesis and neuronal differentiation (Scolnick et al., 2008; Bianchi et al., 2017). Our current data suggest that the effect of IGF1 is global, as suggested by the increased dendritic length in IGF1-treated shControl neurons, concordant with the dendritic changes in mutant R841X and restored R841X-C neurons (Zaslavsky et al., 2019). Therefore, a novel drug may be administrated in combination with IGF1 to fully rescue the defect in shSHANK2 neurons.
Hyperactive neuronal migration remains in shSHANK2 neurons after IGF1 treatment. This warrants further investigation of the mechanisms by which SHANK2 knockdown turns off the static signal in neurons. A recent study demonstrates that another SHANK family member, SHANK3, is involved in integrin-extracellular matrix-mediated cell soma motility via the SPN terminal (Lilja et al., 2017). This study suggests an association between SHANK3 and neuronal dynamics, linking the protein to neurodevelopmental diseases. This study also suggests that the expression level of SHANK3 affects the activation-inactivation oscillatory state of integrin, which interacts with the extracellular matrix, thereby modulating the migration and dynamic behavior of cell bodies (Lilja et al., 2017). Collectively, these observations imply a close relationship between SHANK and neuronal motility. Therefore, it is important to identify drugs that rescue the various phenotypes caused by SHANK2 deficiency to advance therapies for neurodevelopmental disorders.
While the present study is the first to demonstrate that SHANK2 knockdown results in aberrant neuronal motility in the early developmental stage in vitro, there are some limitations. For example, the SHANK2 mutation was not derived from autism spectrum disorder or neuropsychiatric patients. Мoreover, except for the relationship between the SHANK protein family and integrin, the mechanisms by which motility is affected by SHANK2 knockdown remain unclear, requiring further study. Мore importantly, the synaptic transmission alterations in glutamatergic SHANK2 knockdown neurons, such as AМPA and NМDA-dependent electrophysiological changes, as well as its reversibility via IGF1 intervention, were not assessed in our study.
In summary, SHANK2 downregulation in iPSC-derived human neurons perturbs neurite outgrowth and neuronal motility, manifested by decreased dendritic length and branch number, and increased somatic speed. IGF1 administration only rescued the morphological defects, and not the change in neuronal motility. Defects in early developmental processes have a wide range of consequences for the timely, coordinated establishment of connectivity of neuronal networks. Therefore, it is crucial to assess the reversibility and specificity of the varied perturbations to restore normal function in SHANK-related neurodevelopmental disorders. Drug candidates should target not only neurogenesis, but also early neuronal migration.
Acknowledgments:We thank our lab “Guangdong-HongKong-Macau Institute of CNS Regeneration” for providing us platform to do scientific researches. We thank Prof. Duan-Qing Pei from Chinese Academy of Sciences for providing the iPSC cell lines used in the study.
Author contributions:Study design, manuscript writing: LLS; performance of cell culture experiment, live cell imaging, neural morphology reconstruction, and data analysis: STC, WJL; co-writing the initial draft of the manuscript: WJL, QPC and WJZ; critical revision of the manuscript for intellectual content: LBZ, KFS. All authors approved the final version of the paper.
Conflicts of interest:The authors declare that there are no conflicts of interest associated with this manuscript.
Financial support:This study was supported by the National Natural Science Foundation of China, No. 81771222 (to LLS); the Natural Science Foundation of Guangdong Province of China, No. 2019A1515011316 (to LLS); the National Key Research and Development Program of China, Stem Cell and Translational Research, No. 2017YFA0105102 (to LLS); the Guangzhou Science and Technology Innovation Development Special Fund Project of China, No. 201804010212 (to LLS); the Program of Introducing Talents of Discipline to Universities of China, No. B14036 (to KFS). The funding sources had no role in study conception and design, data analysis or interpretation, paper writing or deciding to submit this paper for publication.
Institutional review board statement:All experimental procedures and protocols were approved by Laboratory Animal Ethics Committee of Jinan University of China (approval No. 20170228010) on February 28, 2017. The cell experiments were approved by Medical Ethics Committee of the First Affiliated Hospital of Jinan University of China on February 25, 2017. The experimental procedure followed the United States National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996).
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewers:Huanxing Su, University of Macau, China; Chuntao Zhao, Cincinnati Children’s Hospital Medical Center, USA.
Additional file:Open peer review reports 1 and 2.

- 中国神经再生研究(英文版)的其它文章
- Activation of β2 adrenergic receptors promotes adult hippocampal neurogenesis
- Neuroprotective mechanisms of DNA methyltransferase in a mouse hippocampal neuronal cell line after hypoxic preconditioning
- FOXO3a as a sensor of unilateral nerve injury in sensory neurons ipsilateral, contralateral and remote to injury
- Lycium barbarum polysaccharides related RAGE and Aβ levels in the retina of mice with acute ocular hypertension and promote maintenance of blood retinal barrier
- Enriched environment enhances histone acetylation of NMDA receptor in the hippocampus and improves cognitive dysfunction in aged mice
- Pentraxin 3 contributes to neurogenesis after traumatic brain injury in mice