
桥粒在肺部疾病中的研究进展
2020-06-01 07:29:04姚蕾王燕姚欣
国际呼吸杂志 2020年10期
姚蕾 王燕 姚欣
南京医科大学第一附属医院呼吸与危重症医学科210029
通信作者:姚欣,Email:yaoxin@njmu.edu.cn
桥粒作为一种重要的细胞间连接方式,主要存在于上皮组织和心肌组织中,对于维持组织的结构及功能具有重要作用。1864年,意大利病理学家Giulio Bizzozero在表皮棘层中发现桥粒结构,当时这些通过光学显微镜发现的微小 结 节 被 命 名 为 “Bizzozero 结 节”。1920 年, “桥 粒(desmosome)”由Josef Schaffer进行命名。该名称来源于古希腊语,“desmo”代表结合或固定,“soma”代表小体。桥粒由钙黏蛋白家族、犰狳家族、血小板溶素家族的相关蛋白参与构成[1]。钙黏蛋白家族主要包括桥粒芯糖蛋白(desmogleins,DSGs)和桥粒芯胶蛋白 (desmocollins,DSCs),人类DSGs有4 种异构体:DSG1、DSG2、DSG3和DSG4。DSCs 则 有3 种 异 构 体:DSC1、DSC2 和DSC3[2]。犰狳家族蛋白包括桥粒斑珠蛋白 (plakoglobin,PKG)、桥粒斑菲素蛋白 (plakophilins,PKPs)、β-连环蛋白 (β-catenin)和p120-连环蛋白等,其中PKPs包括3种同种异构体,PKP1、PKP2和PKP3[3-4]。血小板溶素家族蛋白包括桥粒斑蛋白 (desmoplakin,DSP)、网蛋白、包斑蛋白和周斑蛋白等,DSP是最主要的蛋白[5]。
随着电镜技术的发展,桥粒超微结构和组成得以进一步的明确,从结构上可将桥粒分成细胞外核心区,又称桥粒芯、外部致密斑块和内部致密斑块三部分 (图1)[6]。细胞外核心区位于相邻两层细胞质膜中间,由桥粒钙黏蛋白细胞外的结构域组成,并含有特征性的中间线[7]。桥粒钙黏蛋白是跨膜蛋白,它们可桥接到细胞外空间并嵌入到相邻细胞中,一方面能与来自相邻细胞的相应分子直接结合,构建桥粒结构的组装平台;另一方面,它们本身作为跨膜蛋白,有助于从外向内或从内向外传导相关信号[8]。外部致密斑块除外桥粒钙黏蛋白的细胞质尾部,还包括能与之相结合的犰狳家族和血小板溶素家族蛋白[9-10]。PKG 是犰狳家族蛋白成员,能够直接与桥粒钙黏蛋白的胞质尾部结合[11-12]。血小板溶素家族中的DSP 能够与PKG 以及犰狳家族的另一成员PKPs相结合作用[13]。内部致密斑块由DSP和角蛋白中间丝紧密结合构成[14]。
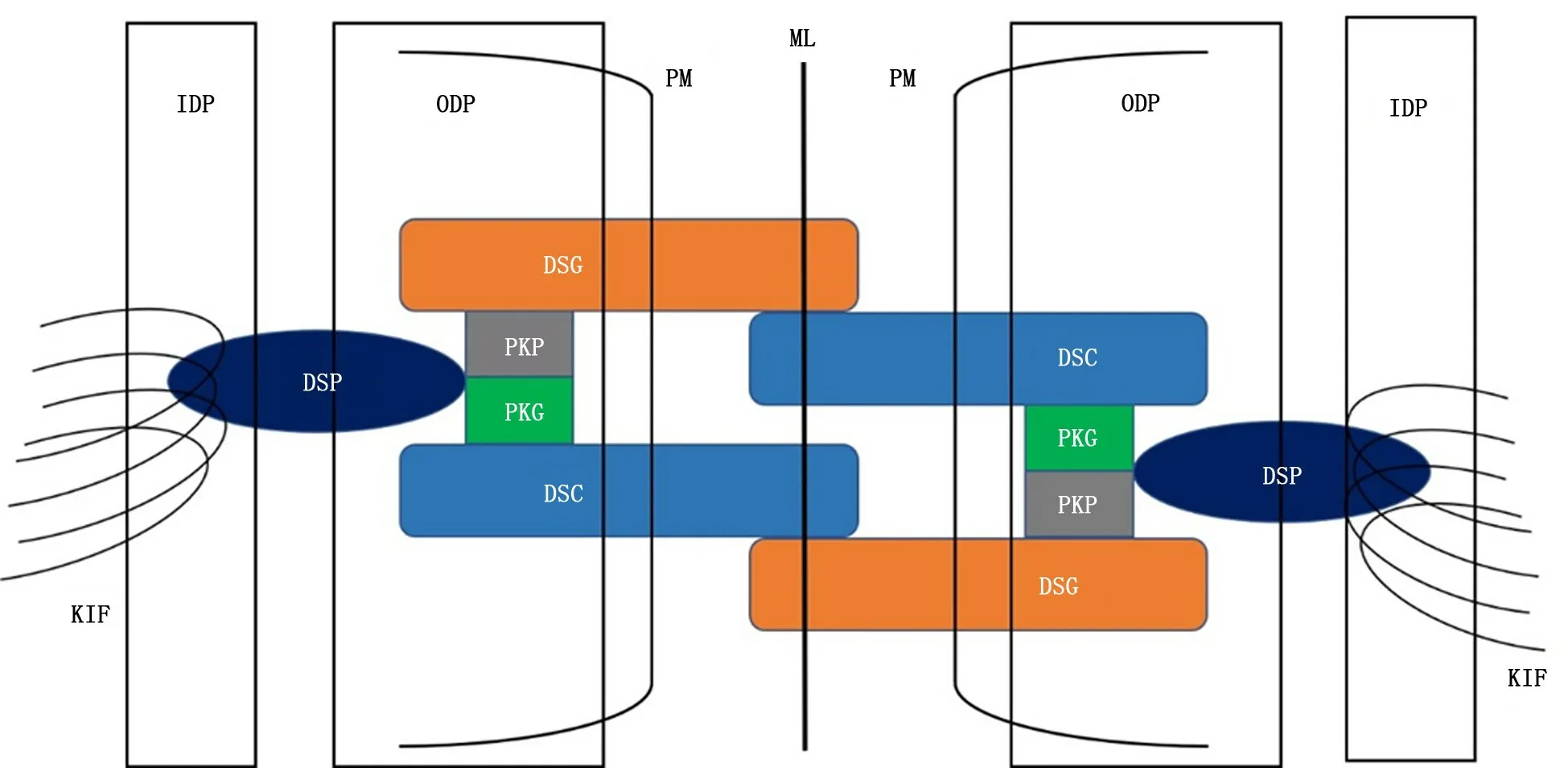
图1 桥粒结构模式图
1 肺癌
研究表明,桥粒可能通过参与细胞增殖、分化、迁移、凋亡及对治疗的敏感性,参与了肺癌的发生[15-17]。
1.1 桥粒蛋白基因在不同病理类型肺癌的表达存在差异 Fukuoka等[18]利用cDNA 表达分析技术证实,DSG3 基因表达水平在肺鳞状细胞癌 (squamous cell carcinoma,SCC)中较高 (强度值>100),而在肺腺癌 (adenocarcinoma,ADC)和正常肺组织中表达极低 (强度值<10)。通过免疫组织化学技术检测肺癌患者肺组织中DSG3的表达情况发现,SCC 阳性率明显高于ADC (79.5%比54.8%,P <0.001)。Savci-Heijink等[19]的免疫组织化学实验结果表明,65例SCC中有64例DSG3 呈阳性,而48 例ADC 中仅1例呈局灶性阳性,该研究所纳入的大细胞肺癌、小细胞肺癌、类癌和恶性间皮瘤病例均为阴性。Saaber等[20]通过组织芯片免疫组织化学技术对原发性肺肿瘤组织的DSG1~3进行分析,发现82例SCC 中有52例DSG2表达阳性,而22例ADC中仅7例DSG2表达阳性 (P =0.009),同时还发现DSG3仅在SCC 患者肺组织中表达,在ADC 中不表达 (P <0.001),且DSG3低表达与较高的肿瘤分级显著相关 (P =0.012)。Gómez-Morales等[21]也证实DSG3免疫组织化学染色对诊断鳞状细胞癌具有高度特异性,同时还发现PKP1 也具有同样作用。Monica等[22]的研究和Tsuta等[23]的研究表明DSC3可作为SCC低分化的标志物,同时也可作为未分化大细胞肺癌鳞状细胞分化的标志物。上述研究表明,桥粒蛋白相关基因表达有望作为鉴别SCC和其它类型肺部肿瘤的辅助生物标志物。
1.2 桥粒蛋白表达与肺癌预后相关 DSGs蛋白低表达与肿瘤分化及进展有关[24-25]。Fukuoka等[18]证实DSG3表达与非小细胞肺癌 (non-small cell lung cancer,NSCLC)患者预后有关,肺肿瘤组织免疫组织化学阴性患者的5年生存率明显低于阳性患者 (20.9%比49.5%,P <0.001)。Cui等[26]对199例原发性肺癌进行免疫组织化学分析,结果表明DSC1的较低蛋白表达与较差的肿瘤分化明显相关(P =0.017),DSC1和DSC3阳性及阴性患者的中位生存期 分 别 为1 158 d 和787 d,1 218 d 和744 d,DSC1 和DSC3表达降低与生存期缩短明显相关 (P =0.045、0.007)。Pantel等[27]的研究表明ADC 患者肺原发肿瘤组织中缺乏PKG 的往往预后不佳。但与之相反的是,在He等[28]的一项关于肺腺癌外科术后的随访研究中,对147例患者肺肿瘤组织进行免疫组织化学分析,结果表明PKG 免疫反应性评分高者 (≥4分)的无病生存期和总生存期更短。另有研究发现PKG 可能是软组织肉瘤肺转移及预后的重要生物标志物,PKG 基因表达水平高者发生肺转移的概率低且5年生存率更高[29]。Furukawa等[30]发现肺肿瘤组织中PKP3的较高表达与肺腺癌患者的不良预后、疾病分期和淋巴结转移有关。
1.3 桥粒参与肿瘤形成的分子机制
1.3.1 钙黏蛋白家族 Saaber等[20]通过RT-PCR 技术检测肺癌细胞系和人支气管上皮细胞中DSG1-3 m RNA 的表达,发现与人支气管上皮细胞相比,DSG1-3在多种肺癌细胞系中表达下降。去甲基化剂5-氮杂-2-脱氧胞苷处理后,部分肿瘤细胞系的DSG2和DSG3 m RNA 得以修复,由此推测DSG2和DSG3在肺癌细胞中的下调可能与DNA 甲基化有关。Cai等[31]的研究发现与阴性对照siRNA 相比,DSG2 siRNA 转染的NSCLC细胞系中CDK2的表达显著降低,而肿瘤抑制因子p27蛋白的表达增加,表明DSG2可能通过下调p27及上调CDK2促进肿瘤的发生、发展。Cui等[26]通过RT-PCR 分析DSC1-3 m RNA 的表达,发现与人支气管上皮细胞相比,其在多种肺癌细胞系中表达下降,且在用不同浓度的5-氮杂-2-脱氧胞苷处理细胞后,部分肿瘤系细胞中DSC1-3 恢复了表达,表明DSC1-3 下调与DNA 甲基化相关。他们进一步研究发现DSC3 通过抑制EGFR/ERK 通路而具有肿瘤抑制的功能,吉非替尼通过抑制EGFR 增加了DSC3的表达。该研究还发现肺癌细胞中DSC3的表达受p53 的调节,且与p53 结合位点中DSC3 DNA 的甲基化有关[32]。
1.3.2 PKG Sechler等[33]研究证实肝细胞生长因子激活物抑制因子1 能够调节细胞迁移并增加NSCLC 细胞对c-MET抑制剂的敏感性。他们进一步研究发现肝细胞生长因子激活物抑制因子1是PKG 的主要新型下游效应物,且PKG 在NSCLC中的抗增殖和抗迁移作用主要由p53诱导。用DNA 甲基转移酶抑制剂5-氮杂-2-脱氧胞苷处理SCLC细胞系H157后能够诱导PKG 的表达并减少肿瘤细胞的增殖。
1.3.3 DSP Yang等[34]研究发现与人支气管上皮细胞相比,DSP在多种肺癌细胞系中表达下降,且在NSCLC 细胞系H157中过表达DSP后,细胞增殖、迁移和侵袭能力均受到抑制,并增加了由吉西他滨诱导的细胞凋亡的敏感性。此外,DSP 过表达增强了PKG 表达,促使T 细胞因子/淋巴增强因子依赖性转录活性降低,Wnt/β-catenin靶基因Axin2和基质金属蛋白酶MMP14表达也降低。小干扰敲除NSCLC细胞系H2228的DSP 后,PKG 表达降低,β-catenin及MMP14表达增加。
为了深入分析ADC和SCC之间差异基因的表达,Hao等[35]对大样本肺癌数据库-GSE43580进行分析,发现了17个细胞连接相关基因在ADC 和SCC 中表达有差异。并通过Gene ontology 分析筛选出8 个桥粒基因:DSC1、DSG1、DSC2、PKG、DSP、PKP1、DSC3 和DSG3,且在人肺癌组织标本中,桥粒基因在SCC 中高表达,在ADC中表达较低。且在ADC 中,而非在SCC 中,桥粒基因的表达与SOX 家族的转录调节因子SOX30表达呈正相关性。SOX30在ADC 细胞系 (A549 和LTEP-a-2)中过表达后能上调桥粒基因的表达,且SOX30作为桥粒基因的关键转录调节因子,可直接结合桥粒基因启动子区ACAAT 序列并激活它们在ADC 中的转录。该研究还发现,miRNA 敲除ADC 细胞系中DSP、PKG、DSC3 的表达可明显减弱SOX30对ADC细胞增殖、迁移和侵袭的抑制作用。与野生型小鼠相比,SOX30 敲除鼠肺组织中DSP、PKG、DSC3的m RNA 和蛋白表达水平下降,SOX30 通过激活Wnt和ERK 信号通路,抑制桥粒相关基因的表达从而促进肺癌的发生发展。上述实验结果为桥粒基因调控提供了潜在的新机制,并为SOX30参与ADC 生长和转移的分子机制提供了研究新思路。
2 特发性肺纤维化 (idiopathic pulmonary fibrosis,IPF)
IPF是一种没有明确病因的慢性进行性肺纤维化疾病。肺泡上皮细胞损伤可能是纤维化肺修复的起始因素,随后引起间充质细胞的迁移、增殖和活化,形成成纤维细胞/肌纤维母细胞灶,导致细胞外基质的过度沉积,肺实质发生不可逆的破坏[36]。Matthes等[37]研究发现IPF患者的肺成纤维细胞PKG 表达水平较健康对照者低,但用siRNA 敲降肺成纤维细胞表达的PKG 后,其特征性成纤维细胞表型(黏附、增殖和凋亡)并未发生改变,因此,尚需进一步研究以明确这些细胞中PKG 减少所致的功能改变。
一项纳入了40例IPF和40名健康对照者的研究发现,在40%IPF患者的血清和肺泡灌洗液中检测到抗周斑蛋白(periplakin,PPL)抗体阳性 (在健康者中均未检测到)[38]。随后Besnard等[39]对PPL 在博来霉素诱导的肺纤维化小鼠模型中的肺损伤和重塑作用进行了深入探究,研究发现博来霉素诱导损伤后,小鼠肺中PPL 表达明显下降。PPL基因敲除鼠与对照小鼠相比,博来霉素诱导损伤后,小鼠生存率明显提高,并且肺纤维化进展得以减缓,其主要可能作用机制是PPL 敲除后促进抗炎细胞因子表达,并改变肺泡上皮细胞的信号通路的转导,包括STAT3的活化和TGF-β/Smad途径的激活。
3 硅沉着病
吸入二氧化硅粉尘是硅沉着病发生的原因,但并非所有在任何特定时间硅尘暴露中的人都会发生硅沉着病,这可能与硅沉着病的遗传易感性有关。Wang等[40]首次证实了DSP基因多态性与中国人群硅沉着病易感性的关系,DSP基因外显子15中的rs2076304与汉族人群硅沉着病风险增加显著相关 (OR =1.53,95%CI:1.03~2.29,P=0.036),进一步功能学研究表明,rs2076304 可能影响RHOXF1的结合。Mathai等[41]研究发现DSP 中的序列变异与IPF相关,rs2076295 基因型与肺组织中DSP 的差异性表达有关。他们对334例IPF 患者和201名健康受试者肺组织进行DSP基因表达测定,结果表明,与对照肺组织相比,IPF患者肺组织中DSP 基因表达量增加了2.3倍(95%CI:1.91~2.71,P <0.000 1),其表达显著增加并且集中表达在气道上皮细胞中,可能是病变肺组织对损伤的强烈且持续的上皮反应的结果。该研究还发现内含子5变体rs2076295与疾病的显著相关性,它与基因表达降低相关,为该基因在硅沉着病病因学中的作用提供了证据。随后,欧洲的一项IPF全基因组相关性研究验证了DSP基因 [rs2076295,OR =1.44 (1.35~1.54),P =7.81×10-28]在IPF患者和健康人群中存在差异性表达[42]。
4 支气管哮喘 (哮喘)
在支气管上皮细胞-基底细胞间,桥粒是重要的锚定附着结构[43-45]。支气管上皮细胞的脱落,是指柱状上皮从基底细胞剥离,基底细胞部分或完全暴露于吸入的物质,这其中就包括过敏原的侵入,从而更进一步加重炎症反应[46]。对哮喘患者的支气管活检组织进行投射电子显微镜检查,发现桥粒长度相对健康对照减少了30%~40%,因此猜测桥粒的缺少可能是上皮脱落的重要因素[47]。研究证实,在哮喘发生、发展早期,气道上皮屏障功能被破坏和环境中过敏原侵入是其重要现象[48]。在Kampf等[49]研究中,炎症因子肿瘤坏死因子α及γ-干扰素刺激人支气管上皮细胞后,免疫细胞化学染色显示上皮细胞桥粒蛋白表达明显减少。鉴于Pillai等[50]在人支气管上皮细胞中检测到桥粒蛋白DSG2,Zuckerman等[51]进一步研究发现,炎症因子肿瘤坏死因子α和IL-13 刺激人支气管上皮细胞后,其DSG2表达下降,且IL-13能诱导DSG2蛋白的裂解。由此,DSG2可能在气道炎症疾病中发挥重要作用,但其具体作用机制仍需进一步研究。另有研究发现PPL 在气道上皮细胞中也有表达,且PPL作为中间丝骨架结构和桥粒斑块之间的连接体,参与上皮细胞聚集、细胞内信号传导和抗原呈递等[52]。Taille等[53]对260 例哮喘患者及40 名健康对照的血清抗-PPL 抗体进行检测,发现18%的哮喘患者抗-PPL IgG 抗体阳性,而在健康对照者中未检测到。另外,在138 例受试者中有12 例 (8.7%)检测到抗-PPL IgE抗体,并且在合并有鼻息肉患者中的阳性率更高,该研究尚不能明确抗-PPL自身免疫是否真正参与哮喘的发病机制,但表明自身免疫在哮喘发病机制中具有重要作用及其作为哮喘治疗靶点的潜在可能。
5 总结
桥粒作为一种结构蛋白,对维持上皮组织的完整性具有重要作用。桥粒家族蛋白还具有其它丰富的生物学功能,研究表明其参与肺部肿瘤、特发性肺纤维化、硅沉着病、哮喘等多种肺部疾病的发生、发展。相信随着研究的深入,桥粒蛋白的临床价值将得到进一步的提升,为肺部疾病的机制、临床诊断、治疗和预后提供研究新思路。
利益冲突 所有作者均声明不存在利益冲突
(收稿日期:2019-08-03)
猜你喜欢
保健医苑(2023年2期)2023-03-15 09:03:04
保健医苑(2022年1期)2022-08-30 08:40:04
中国临床医学影像杂志(2022年2期)2022-05-25 13:24:34
基层中医药(2021年12期)2021-06-05 06:56:36
山东医药(2015年14期)2016-01-12 00:39:43
医学研究杂志(2015年12期)2015-06-10 06:57:46
江苏大学学报(医学版)(2015年2期)2015-04-17 06:49:51
中国医药导报(2015年26期)2015-02-28 22:07:44
郑州大学学报(医学版)(2015年1期)2015-02-27 14:50:26
郑州大学学报(医学版)(2015年1期)2015-02-27 14:50:26
