
含氯挥发性有机物废气在CeO2基催化剂上的低温催化燃烧净化:从高活性到高稳定性再到高选择性
2020-05-29 09:35:42戴启广王幸宜
工业催化 2020年4期
戴启广,王幸宜
(华东理工大学化学与分子工程学院工业催化研究所,上海 200237)
随着工业水平的发展,环境污染问题日益突出,尤其是大气污染问题近年来倍受人们的关注。随着我国“大气十条”的发布、“美丽中国”概念的提出、《打赢蓝天保卫战三年行动计划》的实施、《排污许可管理办法》的出台、生态环境部的组建、《环境保护税法》的实施等举措,意味着国家对治理大气污染高度关注并展现出了坚定决心。挥发性有机化合物(Volatile Organic Compounds,VOCs),通常指常压下沸点低于250 ℃的易挥发有机化合物,包含芳香烃类(三苯、多环芳烃等)、含氧类(醇、酮、酚、酯等)、烃类(烷烃、烯烃等)和含杂原子类(含卤、氮、硫杂原子化合物)等有机物,是造成雾霾的主要前驱体,也是我国严格控制和亟待解决的大气污染源之一。其中含氯有机化合物(Chlorinated Volatile Organic Compounds,CVOCs),也包括其它含卤素有机化合物,由于本身高毒性、高稳定性、难降解等特点成为关注的重中之重。如,在1977年美国公布的129种环境优先污染物中有60种为卤代烃及其衍生物,并被美国国家环境保护局列入了“减少排放的高毒害化学品(共17类)”;欧共体公布的废气“黑名单”上排在首位的是卤代物及其衍生物;我国1989年提出的68种优先控制污染物中有25种为含氯有机物;联合国环境项目国际条约中有12 个被列为首位持久性有机污染物均为含氯有机化合物;2017年我国环境保护部发布的《优先控制化学品名录(第一批)》中有9种含氯/卤素的优先控制化学品;2019年生态环境部、卫健委共同制定颁布的《有毒有害大气污染物名录(第一批)》中涉及的6种有机物中有4种是含氯有机物。然而,目前诸多行业如精对苯二甲酸行业(溴甲烷)、氯碱化工行业(氯乙烯、氯乙烷)、精细化工/制药行业(如聚碳酸酯合成、青霉素生产中存在二氯甲烷)、焚烧/冶炼/烧结行业(二噁英)都存在含氯有机物的使用或排放,更为关键的是在短期内不可能从源头上完全杜绝含氯有机物的使用或排放。为此,我国诸多排放标准中都对含氯/卤有机物制定了严格的排放阈值,《烧碱、聚氯乙烯工业污染物排放标准(GB 15581-2016)》中将氯乙烯和二氯乙烷排放浓度限定在10 mg·m-3和5 mg·m-3之内,《合成树脂工业污染物排放标准(GB 31572-2015)》将二氯甲烷、氯苯类及环氧氯丙烷控制在100 mg·m-3、50 mg·m-3和20 mg·m-3之内,《石油化学工业污染物排放标准(GB 31571-2015)》中对氯甲烷(20 mg·m-3)、二氯甲烷(100 mg·m-3)、三氯甲烷(50 mg·m-3)、四氯化碳(20 mg·m-3)、1,2-二氯乙烷(1 mg·m-3)、氯甲基甲醚(0.05 mg·m-3)、二氯甲基醚(0.05 mg·m-3)、1,2-二氯丙烷(100 mg·m-3)、溴甲烷(20 mg·m-3)、三氯乙烯(1 mg·m-3)、四氯乙烯(100 mg·m-3)、氯萘(5 mg·m-3)、氯乙酸(20 mg·m-3)等诸多含氯有机物都有严格的排放限值,《生活垃圾焚烧污染控制标准(GB 18485-2014)》中更是将二噁英类污染物排放限值限定为0.1 ngTEQ·NM-3。
面对这些严格的排放标准,目前主要是通过后处理技术如吸附、吸收、直接焚烧、光催化降解、催化燃烧等将排放的含氯有机物加以回收或彻底消除。催化燃烧技术作为一种节能、高效、通用的VOCs净化技术被广泛研究和应用,在CVOCs的净化中也备受关注。如欧洲催化研究集群(European Cluster on Catalysis)发布的《欧洲催化科学与技术路线图》多年来持续提出通过催化燃烧方式解决CVOCs排放是一种可行的技术路线,并指出催化剂的开发是其关键所在。通常用于CVOCs催化燃烧的催化剂包括贵金属催化剂(Pt、Pd、Ru、Rh等)、固体酸催化剂(H型分子筛)和过渡金属氧化物催化剂(Fe、V、Cr、Mn、Co、Ni等金属氧化物或复合氧化物)等,近年来过渡金属催化剂由于对CVOCs的高活性、高抗氯中毒能力且价格低廉而备受学术界和工业界的推崇。但是寻找、设计对CVOCs具有更高活性、高稳定性(耐氯中毒失活)、高选择性(抑制副产物尤其是多氯副产物的生成)甚至是广谱性的催化剂/组分仍然是该领域的奋斗目标,为此众多研究人员做出了大量而深入的研究工作。李灿院士早期率先在国内开展了氯苯类有机物的催化燃烧净化,研究工作主要集中在Mn基催化剂上并提出了MnOClx物种为活性中心的观点[1-3]。后续浙江师范大学罗孟飞、鲁继青研究员等对贵金属Pt、过渡金属Cr基等催化剂开展了持续的研究,并在实际应用中做了大量的前期工作[4-6]。华东理工大学的郭杨龙教授等在氯乙烯废气(针对氯碱行业的废气)催化燃烧的基础研究及工业化方面做了大量的工作,催化剂体系主要集中在钙钛矿、CeO2基和Ru基催化剂上,青岛大学的张传辉教授开展了氯乙烯在钙钛矿催化剂上催化燃烧的深入研究并将其进一步扩展到氯代烷烃的催化燃烧[7-8]。浙江大学的周仁贤教授在CVOCs催化燃烧方面开展了诸多原创性、持续、系统且深入的研究工作,催化剂体系主要为过渡金属、分子筛等,尤其是Cr、Ce、Ti、Nb等改性的催化剂,强调了催化剂酸性与氧化还原性在CVOCs催化燃烧中的协同作用[9-12]。绍兴大学的左树峰、杨鹏等在此领域继续开展了广泛、深入的研究[13-15]。武汉工程大学的刘善堂教授在Mn-Ce基以及其它过渡金属如Cu、Cr等催化剂上氯苯的催化燃烧展开了持续性的研究[16-20]。中国科学院过程工程研究所的朱廷钰、刘霄龙、徐文青等研究了Ru基催化剂(Ru/CeO2和Ru/TiO2等)以及过渡金属氧化物催化剂上CVOCs催化燃烧过程中的副产物分布、催化剂构效关系等[21-24]。兰州化物所的唐志诚课题组在Ru/Mn-Ce基催化剂上CVOCs催化燃烧方面也开展了诸多研究工作,如Mn-Ce形貌、结构在二氯苯催化燃烧中的作用、氯物种移除途径等[25-26]。浙江大学的王海强从催化剂结构/形貌、组分、组合等的设计出发,研究了不同催化剂上二氯甲烷的催化燃烧,考察双金属、构效关系以及催化剂/组分多功能性的协同作用[27-29]。清华大学的李俊华教授从生活垃圾/固废焚烧应用角度入手研究了在V基、Mn基、Ce基等催化剂上NOx和氯代烃的协同催化消除[30-32]。南开大学马小东研究了多氯代苯、氯苯在Ca-Fe催化剂上的脱氯、氧化过程,该催化剂表现出极高的脱氯性能[33-35]。此外,还有众多课题组近年来也在CVOCs催化燃烧上做出了大量且独到的工作,如华中科技大学的郭利民课题组在氧化铈、分子筛等催化剂体系上开展了研究工作[36],北京工业大学的邓积光、刘雨溪教授开展了三维有序大孔催化材料在CVOCs催化燃烧方面的研究[37-38],中南大学唐爱东研究了Ce、Fe改性的Mn基催化剂上的二氯苯催化燃烧[39],广东海洋大学万义玲研究了氯乙烯在TiO2-MnOx催化剂上的催化燃烧[40],南京工业大学陈英文教授考察了Mn-Ce蜂窝陶瓷催化剂上低浓度氯苯的催化消除[41],浙江大学的赵伟荣研究了Mn-Ce-Zr 三组分整体式催化剂上氯苯催化燃烧[42],中国科学院城市环境研究所的贾宏鹏通过Mn-Ce催化剂制备方法的控制提高锰铈间相互作用,发现其在氯苯和传统VOCs的催化燃烧上表现出优异性能[43]。另外,2019年国家自然科学基金优青项目(环境方向)中,华东理工大学的詹望成,浙江大学的翁小乐[44-46]以及西安交通大学的何炽[47-48]三位青年学者均涉足CVOCs催化燃烧净化的研究,他们在高性能CVOCs催化剂的设计与可控制备、催化燃烧过程中副产物形成机制与控制、催化剂产业化等方面做出了诸多原创性、深入的工作。综上可以看出,CVOCs催化燃烧的研究目前已经成为VOCs净化领域中的又一个热点研究方向,大量的人力、物力投入到该领域的研究中来。在众多研究者的努力之下,CVOCs催化燃烧已经或正在取得重大突破,有大量的研究成果需要我们去汇总和整理,为后续的研究提供指导和借鉴。
本文仅对本课题组在氧化铈基催化剂方面的一些工作、思路,如从高活性氧化铈的发现,到解决催化剂失活策略的提出,再到副产物的控制等,做简单的总结以期能为其它体系CVOCs催化剂的设计、研究提供一些可借鉴的启发。我们课题组还开展了CVOCs在Co、Mn、Cr、Ti等氧化物及其复合氧化物、硫酸根促进的Fe2O3和Sn-TiOx等催化剂上的催化燃烧,由于篇幅所限相关进展将不再赘述。
1 高活性CeO2发现及失活机制研究
我们课题组于2003年开始CVOCs催化燃烧催化剂的设计、制备工作,并获得了国家自然基金项目“Pt基/介孔分子筛催化净化氯代烃类废气的研究”(20377012)的资助。在项目执行期间我们“意外”发现纯氧化铈(作为Pt/Ce-MCM-41有序介孔分子筛的参照对比样品)对CVOCs催化燃烧表现出异常高的催化活性。从此开启了氧化铈基催化剂上CVOCs催化燃烧的研究,期间得到了国家自然科学基金委的多次持续资助,如“过渡金属氧化物/CeO2纳米催化剂上二噁英类有机化合物催化消除研究”(20977029)、“铈基纳米材料催化剂上氯代有机污染物的低温催化消除”(21277047)、“疏水性氧化铈纳米材料的制备及其在含氯有机污染物低温催化净化中的应用”(21307033)、“稀土基催化剂上含氧、含氯挥发性化合物低温催化消除研究”(21477036)和“多组分、多尺度修饰的多功能稀土纳米复合材料催化剂上挥发性含氯、含硫、含氮有机化合物催化净化”(21976056),此外,也得到了863、973、国家重点研发计划等的大力资助。在早期的研究中,我们发现通过硝酸铈直接热分解制备的氧化铈对多种CVOCs均表现出非常高的初活性,如图1所示,200 ℃左右氯代烷烃即可达到完全转化,即使是高稳定性的氯代烯烃如三氯乙烯和四氯乙烯在300 ℃下也能获得90%以上的转化率[49]。在2007年第五届国际稀土开发与应用研讨会暨第五届国际稀土学术会议上,国家973稀土催化项目首席科学家卢冠忠教授所做的“镧铈催化剂处理挥发性有机化合物的低温催化氧化”大会报告给予了详细介绍。

图1 不同CVOCs在CeO2上的催化燃烧活性Figure 1 Catalytic combustion of different CVOCs over CeO2 catalyst
随后我们也研究了其它稀土氧化物(商业试剂和硝酸盐直接热分解制备的稀土氧化物),结果发现仅CeO2表现出异常高的活性,Pr和Sm的氧化物也具有一定的活性。另外,我们发现稀土氧化物的制备方法尤为重要,大多数商业稀土氧化物试剂包括CeO2对CVOCs几乎没有活性(试剂性质的氧化物可能是经过高温焙烧的)。但为什么CeO2对CVOCs表现出高活性?活性中心是什么?我们经过对纯氧化铈物化性质的分析和深入认识后认为可以作为CVOCs活性中心的主要包括以下几个方面:(1)表面羟基(Surface hydroxyl groups,孤立的碱性羟基和桥式的酸性羟基),(2)碱性位/碱性晶格氧(Lattice oxygen species,O2-),(3)活性氧物种/表面吸附氧物种(Reactive oxygen species,Oads),(4)氧空穴/缺陷位(Oxygen vacancies,□)以及(5)Lewis酸性位(Ce3+/4+)。首先,很多研究认为大多数过渡金属、Al2O3等催化剂上的表面羟基是CVOCs活化解离的主要活性中心且C—Cl键的活化解离是CVOCs催化燃烧的第一步、速控步骤(C—Cl键相对于C—H键更容易活化),但我们通过氘代氯代烃、D2O-氯代烃、催化剂表面羟基氢氘交换等原位红外以及质谱跟踪的程序升温反应手段发现,尽管原位红外中能观察到羟基的消耗峰,但是这个消耗峰即使在室温下甚至是更低温度也存在(通常催化剂在室温下并没有活性)。与此同时,(2 800~3 600) cm-1范围内出现新的宽峰且随温度的升高而逐渐消失(归属为-O-H···Cl-VOCs,即氢键吸附,为非选择性吸附,这与实验观察到的CVOCs在碱性、酸性羟基上均有吸附现象吻合—CeO2上通常在3 700 cm-1和3 660 cm-1处同时出现负峰),在氘代实验中发现氘代羟基峰(2 710~2 500) cm-1的存在(说明羟基并没有在真正意义上被消耗)等一系列实验现象和结果证明表面羟基作为直接的活性中心可能性非常低,其主要作用在于富集/吸附CVOCs于催化剂表面和调变无机氯产物的选择性(在较高温度下B酸性质的羟基与活化的Cl结合生成HCl)。另外,如果表面羟基是直接的活性中心那么反应机理应该是一个B酸催化途径,但酸催化的CVOCs催化燃烧没有观察到如此低温下进行的,而且通常羟基B酸强度比较弱。氧同位素实验表明氧物种包括晶格氧和活性氧均不是C—Cl键吸附解离的活性位,它们更主要的作用是C—H键活化、含碳碎片的后续氧化。通过多种控制实验以及理论计算,最终我们更倾向于认为氧空穴、Ce3+/4+是C—Cl键吸附、解离的活性中心(带负电荷的Cl更容易在缺电子、带正电荷中心上吸附即正电荷吸附,类似于L碱在L酸上的化学吸附),尤其是后者(推测其它过渡金属也具有相同的活性中心);表面羟基在CVOCs的物理/氢键吸附、HCl选择性方面起主要作用;碱性晶格氧与副产物的产生有密切关系(涉及C—H键活化、氢转移等反应);而表面吸附氧物种是C—Cl键断裂之后含碳碎片继续完全氧化的活性物种。简而言之,氧空穴、Ce3+/4+是主要活性中心(C—Cl键吸附、解离),而表面羟基、晶格氧、活性氧影响CVOCs在CeO2上催化燃烧的产物选择性。
然而,在后续研究中我们发现纯氧化铈虽然活性极高但却极易失活,如图2所示,在短短的几个小时甚至是十几分钟后三氯乙烯的转化率便快速下降且低温下更容易失活[50]。通常对于VOCs催化燃烧催化剂的失活原因主要有积炭失活、催化剂结构变化、中毒失活等,通过对反应前后纯氧化铈性质的研究(TG、TPO、元素分析、Raman/XRD、XPS等)发现氧化铈在反应后并不存在积炭,其结构/价态等均未发生明显的变化,但表面积累了大量的无机氯物种,由此推断氧化铈的失活来自于氯中毒。由于氧化铈结构/价态在反应前后并未变化,可以排除氧化铈被部分或完全氯化而氯中毒失活的可能性,进一步通过一系列的控制实验(失活催化剂焙烧再生、水蒸气吹扫再生、Cl-可控吸附等以及理论计算)发现无机氯物种(Cl-)的简单吸附即可导致氧化铈的失活,即活性位被无机氯物种占据是纯氧化铈失活的主要原因。这主要是由于Ce3+/4+对Cl物种具有较强的吸附能力,也正因为如此才对CVOCs表现出极高的活性(强解离C—Cl键的能力),从而导致无机氯物种不能及时脱离活性中心,理论计算结果也给予了验证(即使反应生成了HCl,也会很容易在CeO2上再次被解离、吸附,但有意思的是CeO2对Cl2的吸附、解离能力则不强)。另外,在后续研究中我们合成了不同形貌、不同暴露晶面的氧化铈如氧化铈纳米棒[(110)和(100)晶面)、氧化铈纳米立方体[(100晶面]和氧化铈纳米八面体[(111)晶面],由于纳米棒暴露较多的高能面、较多的氧空穴、较好的储放氧能力对CVOCs表现出最高活性(C—Cl键解离),但由于高能、多缺陷的晶面对无机氯物种的吸附也更强从而导致了更快的失活[51]。

图2 三氯乙烯在CeO2上催化燃烧的稳定性Figure 2 Stability of CeO2 catalyst for catalytic combustion of trichloroethylene
2 CeO2失活的解决策略
我们提出了“两种途径三种机理”解决CeO2的快速失活问题[52],如图3所示,通过B酸中心引入、氧氯交换和Semi-Deacon Reaction机理将解离吸附的氯物种分别以HCl或Cl2的形式脱离活性中心,采用这些策略合成的CeO2基催化剂对CVOCs催化燃烧表现出高活性的同时也具有较好的稳定性。

图3 提高氧化铈催化剂CVOCs催化燃烧稳定性的策略示意图Figure 3 Strategies of improving stability of CeO2 catalyst for catalytic combustion of CVOCs
2.1 过渡金属掺杂:氧氯交换途径
由上述可知,尽管纯氧化铈对CVOCs催化燃烧表现出极高的初活性,但会因为氯物种的强吸附而快速失活,由此预测可以通过无机氯物种的快速脱离活性中心或抑制吸附来解决CeO2失活问题。为此,我们课题组花了大量的精力寻找从活性中心上快速移除吸附氯物种的方法/途径。由于过渡金属相对于其它类型催化剂具有更好的耐氯中毒能力且通常过渡金属的掺杂会导致氧化铈产生更多缺陷位而有利于活性的提高。因此,Cu、Co、Ni、Fe、Cr、Mn等过渡金属掺杂的氧化铈首先被用于三氯乙烯的催化燃烧,结果如表1所示。由表1可知,过渡金属的掺杂均明显提高了氧化铈的活性,其中Mn的掺杂效果最佳。副产物(如加氯副产物四氯乙烯和脱氯副产物一氯乙炔、二氯乙炔)方面存在明显差异,Cu、Co、Ni的掺杂导致了四氯乙烯生成,在Fe、Co、Ni上观察到了少量的脱氯气/脱HCl副产物,而Mn上没有检测到明显的氯代副产物。另外,Cu、Mn掺杂的CeO2在较低的温度下均表现出较好的稳定性,如图4所示。CuCeO2催化剂上产生的副产物四氯乙烯在低温下不能完全氧化,只有温度高达400 ℃时才能彻底消除。MnCeO2相比其它过渡金属掺杂表现出更好的低温稳定性(250 ℃)以及更少的副产物。多氯副产物的形成来自过渡金属在反应过程中被部分或完全氯化而形成的过渡金属氯化物,而其通常是烷烃或氯代烃多氯化的催化剂(生活垃圾焚烧过程产生二噁英通常归结为由于烟灰中含有Cu、Fe等成分)。

表1 三氯乙烯在不同过渡金属掺杂氧化铈上的催化燃烧
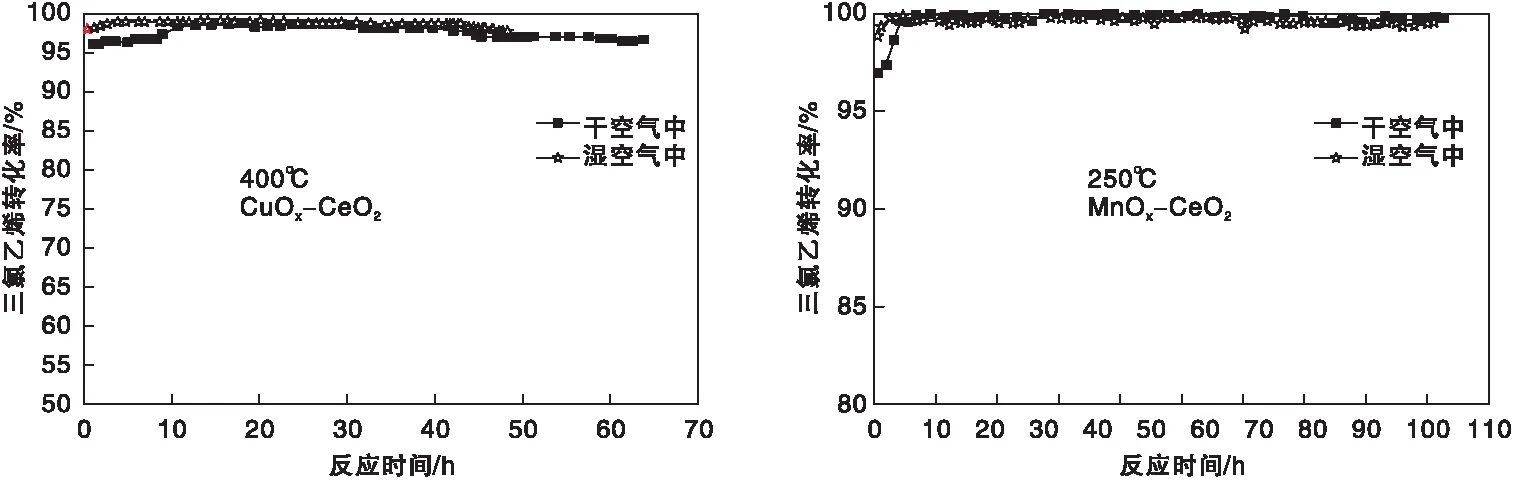
图4 在Cu、Mn掺杂CeO2催化剂上三氯乙烯催化燃烧稳定性Figure 4 Catalytic combustion stability of trichloroethylene over Cu,Mn doped CeO2 catalysts
鉴于MnCeO2催化剂对CVOCs催化燃烧表现出较好的综合性能,我们对其单独进行了深入的研究。结果表明,解离吸附在氧化铈活性中心上的无机氯物种能够通过氯氧交换的方式转移至过渡金属Mn上形成MnOClx(表面Mn物种被部分氯化),由于CeO2具有较好的氧迁移、储放氧性能,在较低温度下能够促进MnOClx被再次氧化,最终氯物种以Cl2形式脱离催化剂表面,氧化铈的活性中心得以再次暴露而部分氯化的Mn则被循环回氧化态,整个C—Cl键解离、氯氧交换循环过程如图5所示。另外,由于Mn多价态特性、优异的氧化还原能力、易氧氯交换(在较低温度下即可进行),所以多氯副产物难以生成。其它过渡金属掺杂的氧化铈同样存在与Mn类似的过程,在此途径中主要利用的是过渡金属的化学不稳定性(越不稳定越容易实现氯物种转移,但也存在更容易生成多氯副产物问题),因此该途径的主要研究方向是通过催化剂制备方法、组分、助剂等的控制使其在较低温度下完成氯氧交换循环、抑制多氯副产物的产生(避开氯代过渡金属催化多氯化反应的适宜温度)。

图5 CVOCs在MnCeO2催化剂上催化燃烧过程中氯氧交换过程Figure 5 The substitution process between chlorine and oxygen in MnCeO2 catalyst for catalytic combustion of CVOCs
2.2 RuOx负载:Semi-Deacon Reaction途径
通过容易氯化的过渡金属氯氧交换途径实现氯物种的快速脱除策略,尽管能够显著提高CeO2的耐氯中毒能力,尤其是MnCeO2催化剂在CVOCs催化燃烧中堪称完美,但在复杂的实际VOCs催化燃烧过程中仍然存在被完全氯化的可能(高浓度CVOCs和水蒸气,低氧浓度、低温等情况),最终导致催化剂的失活和多氯副产物的产生。通过文献调研,我们发现RuOx广泛用于HCl氧化制氯气过程(Deacon Reaction),能将HCl解离和氧化为Cl2,表现出高的活性和化学稳定性(在高浓度HCl和Cl2存在下具有高稳定性)[53]。我们前期的研究发现Cl2不容易吸附在CeO2活性中心上、不会导致其失活,而CeO2的失活主要来自解离的Cl自由基和Cl-物种的吸附,另外,解离吸附的氯物种作为活性的氯物种应该比稳定的HCl更容易氧化(不需要解离H—Cl键),因此,我们提出通过Semi-Deacon Reaction途径(即解离吸附在CeO2上的Cl自由基或活性Cl直接氧化成Cl2)在较低温度下将吸附在活性中心上的氯物种转变成Cl2而脱离催化剂表面,从而提高CeO2催化剂稳定性,避免出现过渡金属氧化物被部分或完全氯化的情况。我们的研究发现掺杂有Ru的CeO2(1%Ru-CeO2)催化剂在氯苯的催化燃烧中不仅表现出高的活性(275 ℃时,氯苯的转化率为98%以上),而且具有较高的稳定性,82 h内活性没有任何降低,但遗憾的是该催化剂在较低温度下仍然会快速失活,如图6所示[54]。通过HCl、Cl2定量分析以及质谱跟踪的程序升温反应等手段证实Ru-CeO2催化剂高稳定性的原因在于Ru组分具有较好的Deacon 反应活性,能够将吸附在催化剂活性位上的Cl以Cl2形式转移出催化剂表面,而当温度较低时由于无法高效率地将吸附氯物种以Cl2形式完全脱离催化剂表面因而仍然面临失活可能。因此,通过提高该催化剂Deacon 反应活性预期能够进一步降低获得CVOCs稳定转化的温度。

图6 氯苯在1%Ru-CeO2催化剂上催化燃烧的稳定性Figure 6 Stability of 1%Ru-CeO2 for catalytic combustion of chlorobenzene
对HCl氧化制Cl2工业Ru基催化剂的大量研究表明高分散的Ru以及Ru负载于特定载体如金红石型的TiO2上会表现出更好的Deacon反应活性(更低温度下HCl氧化)[55]。为此,我们考察了Ru/CeO2和Ru/Ti-CeO2催化剂上氯苯的催化燃烧[56],结果表明,Ru高分散的Ru/CeO2相较于Ru-CeO2具有更好的低温稳定性,在250 ℃下氯苯即可达到稳定转化,如图7所示,但由于Ru占据了CeO2的部分活性中心以及缺陷位的减少导致其活性略有降低;当Ru负载于Ti掺杂的CeO2后氯苯的稳定转化温度进一步降低至200 ℃。这些结果完全符合我们的预期,即提高Deacon 反应活性可以在更低温度下获得CVOCs的稳定转化,与此同时也可极大程度上抑制多氯副产物的形成。

图7 不同温度下氯苯在1%Ru/Ti-CeO2和1%Ru/CeO2催化剂上的稳定性Figure 7 Stability of 1%Ru/Ti-CeO2 and 1%Ru/CeO2 for catalytic combustion of chlorobenzene at different temperatures
此外,通过Semi-Deacon反应途径移除吸附氯物种的策略,我们推测并证实了纯氧化铈在较高温度下如(330~350) ℃以上对于CVOCs的催化燃烧也具有较好的稳定性[56],也就是说纯CeO2同样具有Deacon反应活性,后来这也被华东理工大学的郭杨龙教授采用氧化铈用于Deacon反应的系列报道所验证[57]。CeO2、Ru-CeO2、Ru/CeO2及Ru/Ti-CeO2上氯苯催化燃烧稳定转化的最低反应温度如图8所示。

图8 CeO2、Ru-CeO2、Ru/CeO2及Ru/Ti-CeO2上氯苯催化燃烧稳定转化的最低反应温度Figure 8 The lowest temperature for stable catalytic combustion of chlorobenzene over CeO2, Ru-CeO2, Ru/CeO2 and Ru/Ti-CeO2 catalysts
根据催化反应的基础理论氧化铈用于CVOCs催化燃烧的“逆反应过程”烷烃氯氧化也应该具有可行性,厦门大学的王野教授在甲烷氧氯化制氯代烃相关系列工作中也给予了证实[58],同时我们也推测Ru/CeO2、Ru/Ti-CeO2在上述两类反应中具有更好的性能。另外,最新一代Deacon反应的Ru基催化剂采用Al2O3稳定的SnO2作为载体,表现出更好的低温HCl氧化性能[59],为此我们采用Sn掺杂的CeO2负载Ru用于氯苯的催化燃烧,初步结果验证了Ru/Sn-CeO2催化剂相较于Ru/Ti-CeO2具有更好的活性和低温稳定性,原因在于SnO2具有较好的氧化还原性以及Ru/SnO2更好的Deacon反应活性。该催化剂的核心在于SnO2的高度分散和稳定化(SnO2较差的化学稳定性,需利用CeO2稳定),以及Ru在SnO2上的定向锚定(提高利用效率)。另外,我们发现Ru、Ti、Sn均具有相同结构(即金红石结构)且与CeO2结构非常接近,根据广泛存在于负载型催化剂中的相似相分散现象(“Like-Disperses-Like”,负载金属氧化物和载体的晶型相似时,可有效促使M-O-S界面氧键的形成,提高被负载物的单层分散阈值)[60], 我们预测具有金红石结构的Mn、Mo、Cr、V等掺杂的氧化铈作为Ru的载体在CVOCs催化燃烧中应该也表现出较好的低温稳定性。
总之,利用具有较好化学稳定性Ru的修饰、通过Semi-Deacon反应途径提高CeO2在CVOCs催化燃烧中的稳定性具有可行性,寻找能够在更低温度下氧化解离吸附的氯物种以降低多氯副产物产生的组分仍然是研究的方向之一,通过诸如Ti、Sn、Mn、Cr或Mo等第三组分的掺杂改性可以部分实现这一目标,由于Ti、Mo相较于Sn、Cr、Mn具有更好的化学稳定性,被认为是更好的CeO2改性组分,后几者则需解决化学稳定性的问题(抑制部分或完全氯化)。
2.3 B酸中心引入:HCl途径
CVOCs催化燃烧催化剂除了应该具有较好的活性和稳定性外,产物的选择性尤其是抑制更具毒性的多氯副产物的形成也尤为关键,理想的CVOCs催化燃烧反应应该是CVOCs+O2→CO2+H2O+HCl。但是通过氧氯交换或Semi-Deacon 反应策略提高CeO2稳定性时,氯物种被完全氧化为具有高反应性、氧化性、腐蚀性的Cl2,易导致多氯副产物形成(尤其是在金属元素存在下),此外Cl2的产生对反应器材质也提出了更高的要求。因此,抑制Cl2产生提高HCl选择性也是CVOCs催化燃烧催化剂研发过程中首要考虑的问题之一。在氧氯交换和Semi-Deacon Reaction途径中不难发现Cl2的产生均跟催化剂的氧化能力有直接的关系,如果引入B酸性位抑制氯物种深度氧化同时提供H+提高HCl选择性,预期能够改善产物选择性。另外,大量的研究也表明固体酸催化剂在CVOCs催化燃烧中具有较好的抗氯中毒能力,氯物种主要以HCl的形式脱离反应体系。众所周知,VOx作为典型的两性氧化物既具有一定的氧化能力(晶格氧,选择性氧化)又具有丰富的、可以作为B酸性位的羟基,广泛用于选择性氧化、垃圾焚烧、脱硝、硫酸生产等过程,尤其是VOx还具有优异的抗氯中毒能力和化学稳定性。我们的研究表明负载VOx能够显著提高CeO2的稳定性,主要原因在于VOx上丰富的B酸性位使CeO2活性位上的氯物种以HCl形式形成于VOx物种上,避免了在氧化铈活性中心上的累积。通过此途径提高氧化铈稳定性一个显著优点是对反应温度的不敏感性(稳定性的提高不依赖于反应温度),即VOx/CeO2催化剂在较低温度下如150 ℃也具有较好的稳定性,如图9所示[61-62]。由于没有高反应性的Cl2形成以及VOx的化学稳定性,反应过程中并无多氯副产物的产生。但由于氧化能力的削弱、脱氢性能的增强(晶格氧催化),VOx/CeO2会产生非完全氧化的副产物如CO和脱氯有机副产物。因此,提高完全氧化产物的选择性是该催化剂研究的方向之一,另外VOx物种具有生物毒性也制约了该催化剂的实际应用。

图9 反应温度对6.0%VOx/CeO2催化剂活性的影响Figure 9 Effects of reaction temperature on activity of 6.0%VOx/CeO2 catalyst
H型分子筛类固体酸催化剂具有较好的CVOCs催化燃烧活性和抗氯中毒能力且多氯副产物较少,但由于氧化能力较弱及酸性较强而容易产生积炭失活。大量的工作集中在采用过渡金属负载改性提高其氧化能力,但是由于过渡金属暴露在外表面导致氯中毒失活和多氯副产物的产生,尤其是CeO2改性时反应过程产生的无机氯物种仍然容易在CeO2上吸附、累积且氯吸附后的CeO2氧化能力也将大大降低而不能完全解决积炭问题。由于ZSM-5等分子筛具有较好的成膜特性(分子筛膜),我们从纳米材料合成角度设计了HZSM-5包覆的CeO2复合纳米材料(CeO2@HZSM-5)并用于二氯乙烷的催化燃烧[63],以期解决ZSM-5积炭失活、抑制多氯副产物形成、避免具有生物毒性的VOx使用。实验结果表明,CeO2@HZSM-5用于二氯乙烷催化燃烧具有较好的稳定性并未观察到明显的积炭、氯中毒失活现象,与此同时也未检测到多氯副产物,如图10所示。CeO2@HZSM-5尽管在稳定性、副产物方面表现出优异的性能,但是该催化剂活性相对较差,如二氯乙烷在350 ℃以上才能完全转化,与传统的分子筛固体酸催化剂接近。由此可见CeO2在此中并未起到CVOCs活化的作用,原因在于致密的ZSM-5晶体对氧化铈活性位的覆盖以及反应物分子扩散受限。此外,CeO2@HZSM-5在抗水能力方面具有一定优势,可以通过简单的Si/Al比调节控制催化剂的亲疏水能力从而优化CVOCs吸附能力、抗水性能,未来还可通过纳米或片状ZSM-5的包覆,解决CVOCs扩散至氧化铈活性位的路径以提高活性。除了采用传统固体酸分子筛来替代具有生物毒性的VOx之外,我们发现通常作为VOx/TiO2工业催化剂助剂的Mo和W同样具有较好的化学稳定性、抗氯中毒能力以及丰富的酸中心,而在CVOCs催化燃烧催化剂VOx/CeO2中VOx不被认为是主要的活性中心,因此我们采用了环境更加友好的Mo或W取代V负载于CeO2上,实验结果证实了Mo和W具有与V一样的效果,可以明显提高CeO2的稳定性以及抑制多氯副产物的产生,但是非完全氧化产物仍然不可避免,是该策略需要解决的难题。

图10 二氯乙烷在CeO2@HZSM-5 上的稳定性和稳定性测试后催化剂的程序升温氧化曲线Figure 10 Catalytic combustion of dichloroethane over CeO2@HZSM-5 and TPO curves of spent catalyst
3 稳定性与选择性的兼顾策略
由上述可知,采用氧氯交换、Semi-Deacon Reaction和B酸中心引入途径均能显著提高CeO2的稳定性但存在明显的差异,前两者中氯最终主要以Cl2形式移除而造成多氯副产物的形成,而后者以HCl为主是较为理想的产物,不会导致多氯副产物产生但非完全氧化产物不可避免。基于副产物毒性、催化剂化学稳定性等的综合考虑,我们认为B酸性中心引入是一个更好的途径,重点在于适当增强氧化能力。对于抑制多氯副产物产生(引入B酸性位)的同时提高完全氧化产物选择性的问题,我们认为可以通过两个途径加以解决:(1)通过过渡金属元素的掺杂提高V、Mo、W或ZSM-5/CeO2的氧化能力,(2)引入不会显著降低CeO2氧化能力的B酸中心(研究表明CeO2自身的氧化能力足以获得完全氧化的产物如CO2)。
我们首先考察了第一种方案,通过溶剂热法制备了Fe掺杂的CeO2纳米片再负载VOx、MoOx或RuOx等,结果表明,催化剂对二氯乙烷表现出较高活性以及低温稳定性,且无非完全氧化产物生成,尤其是CO的选择性几乎为0,如图11所示[64]。另外,其它过渡金属如Mn、Co、Cu等的掺杂也会有相同的效果,具有普适性。但是过渡金属的掺杂仍然会面临在苛刻条件下如高浓度CVOCs、高水蒸气或贫氧条件下被部分氯化,从而导致氧化能力降低和多氯副产物产生。

图11 二氯乙烷在V、Mo、Ru负载的Fe-CeO2上催化燃烧的稳定性和选择性Figure 11 Stability and selectivity of dichloroethane catalytic combustion of over V, Mo and Ru supported Fe-CeO2
通过第二种方案提高完全氧化产物选择性的核心是找到合适的B酸中心或寻找更优的催化剂制备方法,众多研究报道负载于具有丰富表面羟基载体如SiO2、Al2O3、过渡金属氧化物等上的磷酸根表现出B酸性、高化学稳定性并且对载体氧化等性能的影响较弱(通过羟基锚定磷酸根),尤其是采用有机磷作为前驱体时。为此,我们系统研究了不同磷前驱体、磷负载量等改性的CeO2并用于二氯甲烷的催化燃烧[65],结果如图12所示。

图12 二氯甲烷在磷酸根改性的CeO2上催化燃烧的稳定性及反应过程Figure 12 Stability teat and reaction process of catalytic combustion of dichloromethane over phosphate modified CeO2
多种测试结果表明有机磷改性成功赋予CeO2显著的表面B酸性位且CeO2并未被磷酸化而保留了其本身的氧化能力,活性测试表明即使在较低温度下也具有较好的稳定性,属于B酸中心提高稳定性途径。另外,非完全氧化产物CO几乎未检测到尤其是在较高温度下,但有少量的脱氯加氢产物一氯甲烷产生,不过与传统的Al2O3等固体酸催化剂高达20%~40%的选择性相比,磷改性CeO2上一氯甲烷选择性仅0.5%~5%,更为重要的是在二氯甲烷完全氧化之前,如250 ℃,一氯甲烷不再生成或完全氧化转化。总体上说,磷酸根改性的CeO2用于CVOCs的催化燃烧具有非常全面的性能。
鉴于对多种CeO2基催化剂,如过渡金属掺杂的CeO2、VOx/CeO2、MoOx/CeO2、CeO2@ZSM-5、P/CeO2和Ru/CeO2等,在不同CVOCs催化燃烧中的活性、稳定性、选择性以及活性组分安全性、化学稳定性的认识,我们认为P/CeO2和Ru/CeO2均表现出较好的活性和稳定性,但也存在诸多差异(提高CeO2稳定性的途径),在CVOCs催化燃烧催化剂中具有较强的代表性,为此我们对两者进行了详细的比较研究[52]。由于水在VOCs催化燃烧中广泛存在(实际废气中水蒸气经常伴随着VOCs共存、燃烧反应也会生成水),水对于两者活性均有抑制作用且均为可逆影响,但由于P/CeO2表面丰富的B酸特性的羟基以及Ru/CeO2疏水性,水对P/CeO2的影响更为显著。水除了对活性产生影响外,实验发现对含氯副产物的选择性也有明显的影响且两者存在区别,如图13所示。水明显抑制了脱氯副产物一氯甲烷在P/CeO2上的生成,而对Ru/CeO2上多氯副产物却有所促进,原因在于P/CeO2上水吸附于碱性晶格氧导致氢负离子的产生(氢转移)受到抑制,从而减少了一氯甲烷的生成,而对于Ru/CeO2,水对Deacon 反应影响甚微且对氯自由基的产生有利从而导致多氯副产物反而有所增加[66]。
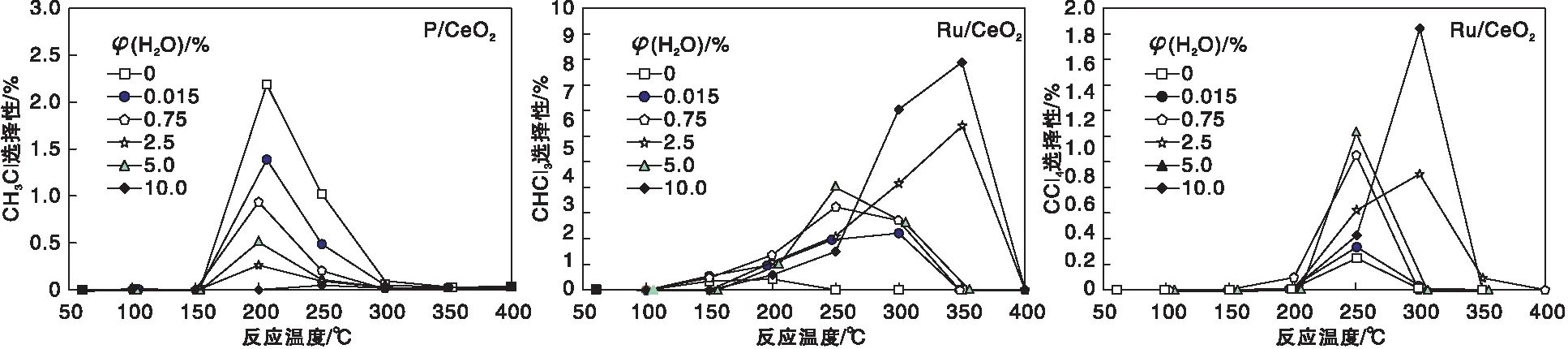
图13 P/CeO2和Ru/CeO2催化剂上水对二氯甲烷催化燃烧过程中含氯副产物的影响Figure 13 Effects of H2O on the formation of by-products during catalytic combustion of dichloromethane over P/CeO2 and Ru/CeO2 catalysts
后续我们也比较了不同类型CVOCs如单碳烷烃二氯甲烷、多碳烷烃二氯乙烷、烯烃氯乙烯、芳香烃氯苯和含氧烷烃氯代环氧丙烷等在P/CeO2和Ru/CeO2上的催化燃烧,结果如图14所示。
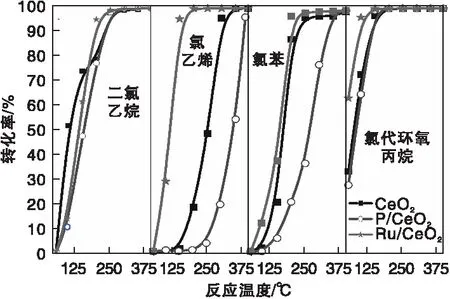
图14 P/CeO2和Ru/CeO2催化剂上不同CVOCs催化燃烧活性Figure 14 Catalytic combustion activity of different CVOCs over P/CeO2 and Ru/CeO2 catalysts
由图14可知,两者均表现出高活性、高稳定性, Ru/CeO2表现出更好的活性,是一个广谱型CVOCs催化燃烧催化剂。不同类型CVOCs催化燃烧多氯副产物易在Ru/CeO2上生成,而在P/CeO2上生成少量的脱氯副产物。
尽管Ru/CeO2和P/CeO2对CVOCs催化燃烧均表现出较好的综合性能(高活性、高稳定性、广谱性),但两者在含氯副产物方面表现出不同的结果甚至具有互补性,因此,我们考察了将RuOx和磷酸根物种分别通过掺杂和负载方式先后引入至CeO2并用于二氯甲烷的催化燃烧,结果如图15所示,P/Ru-CeO2具有与Ru/CeO2和P/CeO2相媲美的甚至更高的活性及稳定性,更为重要的是多氯副产物和非完全氧化产物如一氯甲烷和CO均没有观察到,该催化剂几乎解决了CVOCs催化燃烧过程中的所有难题。另外,由于磷酸根和RuOx均具有较高的化学稳定性,预计该催化剂能够耐受更为复杂、苛刻的反应条件,具有实际应用的前景。

图15 二氯甲烷在P/Ru-CeO2上催化燃烧活性、稳定性和程序升温表面反应Figure 15 Activity,stability and temperature-programmed surface reaction of catalytic combustion of dichloromethane over P/Ru-CeO2
4 结语及展望
(1) 首先发现了对CVOCs催化燃烧具有极高活性的CeO2,通过研究初步确定氧空穴/缺陷位、Ce3+/4+为主要的活性中心,而表面羟基、碱性晶格氧、表面吸附活性氧分别起到CVOCs表面氢键富集、氢转移副产物形成、含碳碎片完全氧化等作用,但是遗憾的是准确且完整的机理仍然不清楚。
(2) 纯的CeO2在CVOCs催化燃烧中面临快速失活问题,CeO2失活的原因在于解离吸附在活性中心的无机氯物种(解离氯或Cl-)无法及时移出导致活性中心被占据而失活,并非传统的积炭和活性组分被氯化而失活。
(3) 提出了“两种途径三种机理”解决CeO2的快速失活问题,通过B酸中心引入、氧氯交换和Semi-Deacon Reaction机理将解离吸附的氯物种分别以HCl或Cl2的形式脱离活性中心,采用这些策略合成的CeO2基催化剂对CVOCs催化燃烧表现出高活性的同时也具有较好的稳定性。但是这些设计的CeO2基催化剂仍然存在一定的问题,尤其是副产物。如,化学不稳定的过渡金属掺杂的CeO2(氧氯交换机理)面临过渡金属被氯化而产生大量多氯副产物问题,高化学稳定性的RuOx负载(Semi-Deacon Reaction机理)由于高反应性Cl2的产生以及表面金属的催化特性同样会导致多氯副产物生成,而B酸中心引入虽然抑制了多氯副产物但由于氧化能力的削弱导致脱氯副产物以及非完全氧化产物CO的产生。综合之下,最终通过CeO2体相掺杂化学稳定的RuOx、表面负载非金属B酸中心(硫酸根、磷酸根等非金属B酸性中心)而设计的P/Ru-CeO2催化剂表现出近乎理想的CVOCs催化燃烧性能(高活性、高稳定性、高选择性)。另外,我们也认为上述所提及的提高CeO2稳定性和选择性的策略同样适用于其它CVOCs催化剂尤其是过渡金属Co、Cu、Mn等氧化物及其复合氧化物催化剂。
经过广大科研人员以及我们课题组15年来的持续研究,国内在CVOCs催化燃烧催化剂的基础研究和深层次认识等方面获得了显著的进展,但距开发出一种真正能够实际应用的催化剂还有一定的差距,无论是在基础研究还是工业示范方面都还存在亟待攻克的各种难题,需要后续持续的研究。
(1) 通过具有特定特性的CeO2可控合成,如高氧缺陷的CeO2量子点、具有限域效应的CeO2纳米管、本征疏水性的CeO2、非金属掺杂的CeO2、表面羟基可控的CeO2以及价态可控且稳定的CeO2等合成,对CeO2高活性的本征来源开展明确、深入的研究,为理性设计更为高效的CVOCs催化燃烧催化剂提供基础性、原理性的指导;
(2) 从活性、稳定性及副产物选择性综合判断,过渡金属如Mn、Co、Cr、Cu等被认为是一类有前景的CVOCs催化燃烧催化剂,但仍需重点解决多氯副产物控制以及活性组分稳定化等问题。此外,通过原位Raman、XPS等手段研究反应过程中催化剂的变化是基础研究的主要方向之一(之前大量工作集中在采用原位红外对反应途径的研究,然而对催化剂的原位研究较少),以明确副产物的形成与催化剂变化直接的关联、寻找稳定化活性组分的方法;
(3) 多氯、脱氯副产物的形成机理和控制途径仍有待深入、全面研究:系统分析、确定各种氯物种的活化、形成(如C-Cl、H-Cl、Cl-Cl键的解离活化,Cl-、Cl自由基等无机氯物种的产生和反应性,以及HCl、Cl2的形成)、理解各种可能的有机反应与副产物间的关联(如加成反应、亲电取代和亲核取代与多氯副产物、消除反应与脱氯副产物、氢转移和歧化反应与脱氯加氢副产物间的关联和决定因素),并由此探索更多途径抑制或二次消除副产物;
(4) 尽管CeO2基及其它过渡金属催化剂对CVOCs表现出广谱性,但对传统的VOCs在活性等方面还有待进一步提高。对于传统VOCs催化燃烧目前工业化最成熟、高效的催化剂仍然是贵金属催化剂,但当存在CVOCs时贵金属则表现出较差的性能(低活性、低稳定性、多副产物),另外,实际工业废气中CVOCs很少单独存在且浓度较低,因此从实用角度出发研究如何提高贵金属催化剂的抗氯中毒失活能力是实现CVOCs催化燃烧工业化应用的最简单、最实用途径。研究如何将CeO2及其提高稳定性、选择性的策略与传统VOCs贵金属催化剂进行复合,完成含有CVOCs的VOCs催化燃烧广谱性实用催化剂开发是未来研究的重点;
(5) 对于CVOCs催化燃烧除了要求催化剂必须具有高的化学稳定性同时,也同样要求其具有高的热稳定性尤其是工业应用的催化剂(工程应用中催化燃烧经常会出现短暂的飞温现象,温度高达650 ℃以上),然而CeO2以及其他过渡金属催化剂的热稳定性较差且在CVOCs催化燃烧研究中并未获得足够关注。因此,在不损失CeO2基等催化剂对CVOCs的高活性情况下,提高催化剂抗热冲击的能力也是未来需攻克的难题之一。
猜你喜欢
英语世界(2023年10期)2023-11-17 09:18:18
保鲜与加工(2021年1期)2021-02-06 06:43:22
科学大众(中学)(2019年3期)2019-05-17 10:04:30
汽车观察(2018年10期)2018-11-06 07:05:26
广东饲料(2016年5期)2016-12-01 03:43:22
中国资源综合利用(2016年12期)2016-01-22 02:02:26
少儿科学周刊·少年版(2015年1期)2015-07-07 17:15:12
河南科技(2015年2期)2015-02-27 14:20:35
应用化工(2014年12期)2014-08-16 13:10:46
食品科学(2013年19期)2013-03-11 18:27:17
