
化学增幅型光刻胶材料研究进展
2020-05-25 00:23:44郑祥飞孙小侠刘敬成穆启道刘晓亚
影像科学与光化学 2020年3期
郑祥飞,孙小侠,刘敬成*,穆启道,刘 仁,刘晓亚
(1. 江南大学 化学与材料工程学院,江苏 无锡 214122; 2. 苏州瑞红电子化学品有限公司,江苏 苏州 215124)
芯片行业涉及到国民经济、国家安全的方方面面,属于战略性行业,2018年我国的芯片进口额更是高达3120多亿美元,远超石油。中兴事件、日韩贸易战也再一次凸显了半导体芯片行业作为国家支柱性产业的重要性。芯片主要是通过光刻、掺杂、刻蚀、封装、测试等工序制作完成[1],根据摩尔定律[2]和Rayleigh公式(R=k1λ/NA)[3],芯片的集成度越来越高,光刻胶的分辨率也随之提高,对应的曝光波长越来越短。光刻技术已从G 线(436 nm)光刻、I 线(365 nm)光刻、深紫外线(deep ultraviolet,DUV)光刻发展到极紫外(extreme ultraviolet,EUV)光刻[4,5],与之对应的光刻胶也随之发生变化。光刻胶中各组分相互作用,影响光刻胶的感度、分辨率、对比度、粘度、粘附性、耐热性、耐蚀刻性等性能[6]。
光刻胶的感度是其性能中的一个重要参数,直接影响到芯片的生产效率和器件制造成本。在光刻领域,量子产率表示光刻胶中的光敏组分吸收一个光子能够引发反应的分子个数,用来表征光化学反应效率。酚醛树脂-重氮萘醌光刻胶(G线、I线胶)中的光敏组分重氮萘醌(diazonaphthoquinone,DNQ)需要吸收3~7个光子才能促使一分子的DNQ发生化学反应,量子产率只有0.15~0.3[5]。显然,需要新型光刻胶来满足其高感度的要求。
光刻胶的分辨率随曝光波长缩短而提高,但高压汞灯于DUV波长处输出的光强度远远低于365 nm、436 nm波长对应的光强度,而且高压汞灯在DUV(254 nm)处释放的光子数量很少[5];准分子激光光源(KrF)激光器输出功率也较低(2019年Gigaphoto 公司KrF 激光器最大输出功率仅50 W),同时也由于半导体工业高产量的需求,更高感度与更高分辨率的光刻胶一直是业内及学术界的研究热点。
1 化学增幅型光刻胶
化学增幅(chemical amplified,CA)概念由Ito、Willson和Fréchet于1982年提出[7],通过链式反应提高光化学反应效率,增强光刻胶感度。化学增幅型光刻胶主要由成膜树脂、光致产酸剂(photochemical acid generator,PAG)、溶解抑制剂、碱性添加剂和溶剂等组成,曝光后PAG分解产酸,催化光刻胶中的酸不稳定(acid labile)基团发生化学反应,催化反应完成后酸又被释放出来,继续催化链式反应[8]。原理图如图1所示。
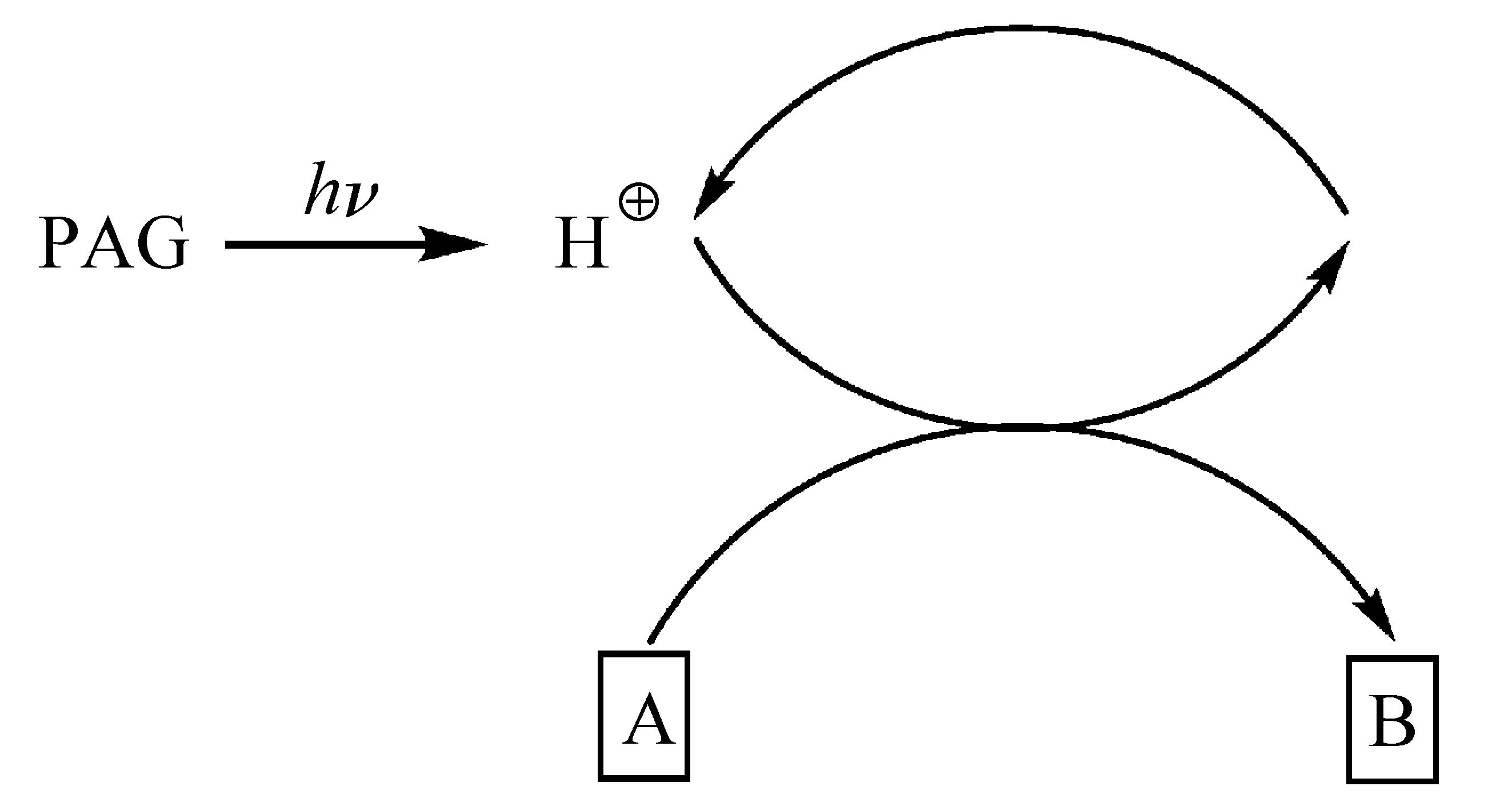
图1 化学增幅型原理示意图[3]
虽然CA概念在80年代被提出,但其机理早在60~70年代便应用于负性光刻胶中,但负胶溶胀效应影响该类光刻胶的分辨率。二十世纪80年代,IBM发明了对-叔丁氧羰基氧苯乙烯基(p-t-butyloxycarbonyl-oxystyrene,PBOCST)光刻胶,用苯甲醚显影,开启了化学增幅型光刻胶(chemical amplified resist,CAR)在集成电路(integrated circuit,IC)领域的应用。随后,通过技术人员的不断开发,新的聚合物结构和反应机理被不断发现和应用[9]。本文根据聚合物的不同反应机理,对CAR聚合物进行了分类阐述。
1.1 脱保护(Deprotection)
聚合物侧链上的酚羟基或羧基的氢原子被碳酸酯、醚或酯等酸不稳定基团取代后,其极性和溶解性发生变化,曝光区域PAG产酸,催化脱保护反应,聚合物侧链重新变回酚羟基或羧基,增加了曝光区域与未曝光区域的溶解性差异。保护基团的热稳定性、脱保护反应的活化能直接影响到光刻胶的性能。该类光刻胶可选择的单体种类多,发生脱保护反应后,聚合物的极性和溶解性发生变化,没有交联结构,可以避免溶胀效应;酸可以连续催化反应,提高光刻胶的感度。酸催化脱保护型CAR是目前DUV光刻领域的主流光刻胶。
(1 ) 碳酸酯:叔-丁氧羰基(t-butlycarbonate,t-BOC)可以作为羟基和氨基的保护基,没有酸催化情况下,t-boc基团加热到190 ℃才能分解(脱保护反应);而酸催化脱保护反应在100 ℃就可以发生,分解生成二氧化碳和异丁烯气体(如图2所示)。根据聚对羟基苯乙烯(poly(hydroxystyrene),PHOST)和PBOCST的溶解性差异,用苯甲醚做显影液,可作为负性光刻胶使用;用四甲基氢氧化铵(tetramethylammonium hydroxide,TMAH)水溶液做显影液,可以作为正性光刻胶使用。为了进一步提高聚合物的耐热性,Ahn等[10]引入t-BOC保护的马来酰亚胺结构,聚合物的玻璃化转变温度超过240 ℃。Novembre等[11]将二氧化硫结构引入到聚合物主链上,制备了单组份光刻胶,在X射线和电子束照射下分解产生硫酸,催化t-BOC进行脱保护反应。六氟异丙醇苯乙烯与对羟基苯乙烯结构具有类似的结构,侧链上的部分羟基用t-BOC保护,其余羟基可与PAG作用,起到抑制溶解的作用,有助于提高光刻胶的性能[12]。此外,聚(羟基苯基甲基丙烯酸酯)[13]、聚(N-羟基苯基甲基丙烯酰胺)[14]等类似结构也有相关研究。异丙基、α-甲基苄基衍生物、1-(2-四氢糠基)乙基等碳酸酯类结构比t-BOC具有更高的热稳定性[14]。图3是部分t-BOC保护的聚合物结构式。
t-BOC在脱保护反应过程中,部分叔丁基阳离子与PHOST发生O—烷基化和C—烷基化反应(如图4所示),这些副反应降低了光刻胶的溶解速率,导致光刻胶对比度降低[15]。
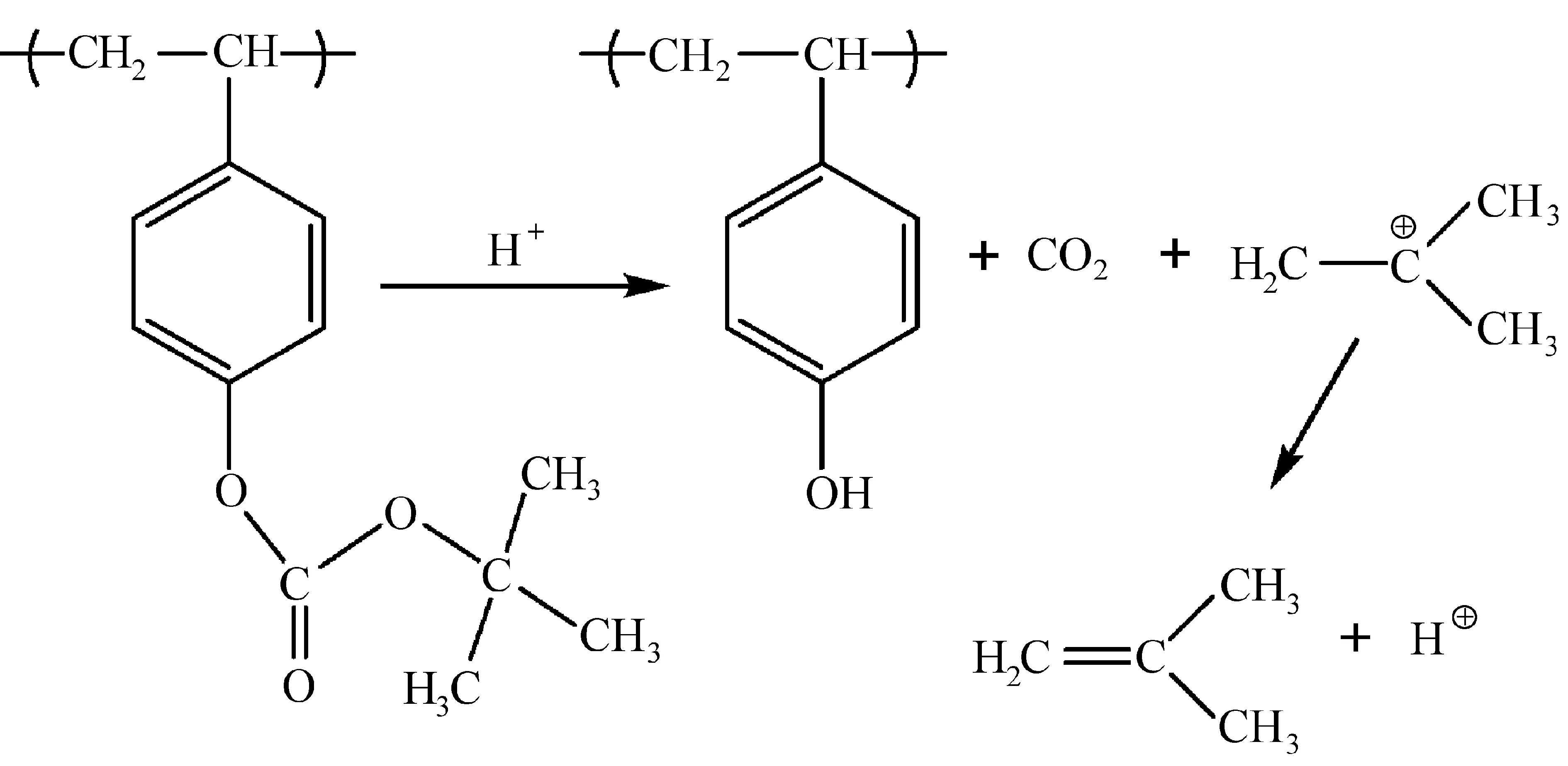
图2 酸催化脱保护反应(PBOCST光刻胶)
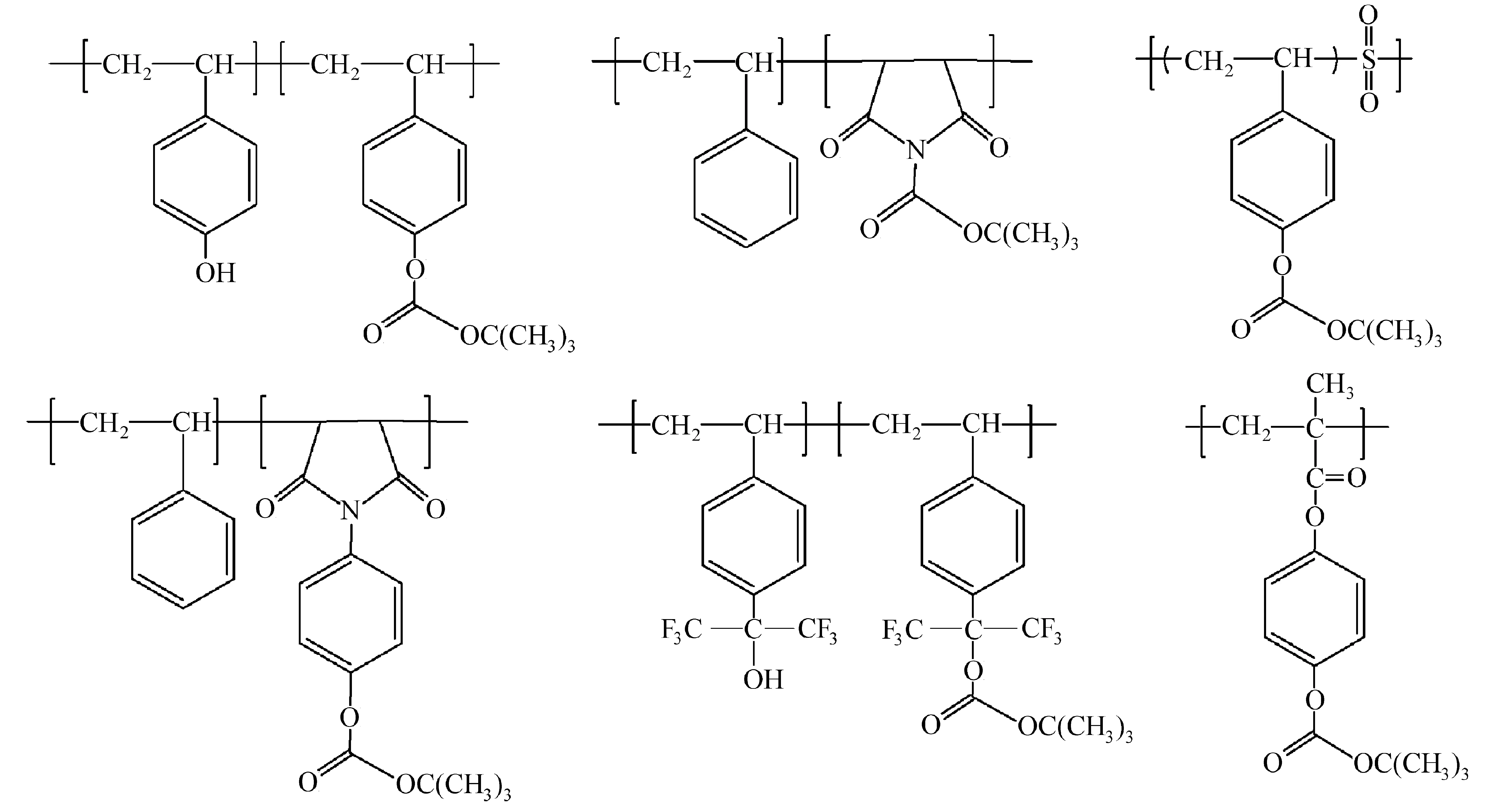
图3 部分t-BOC保护的聚合物结构式[10-14]
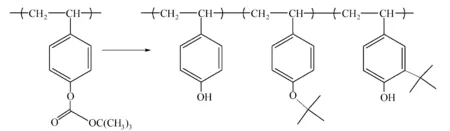
图4 t-BOC脱保护中的副反应[15]
(2) 醚和酯类:PHOST、聚(甲基)丙烯酸、聚(4-乙烯基苯甲酸)的羟基和羧基可以用叔丁基[16]和叔丁氧基羰基甲基[17]等酯或醚基团保护起来,见图5。由于聚(4-乙烯基苯甲酸酯)和聚(4-乙烯基苯甲酸)在248 nm波长处的光密度分别为1.1/μm和 3.4/μm,因此并不适用于DUV光刻胶[18]。IBM开发的ESCAP(environmentally stable chemical amplified photoresist)光刻胶采用对羟基苯乙烯和丙烯酸叔丁酯结构,叔丁基比t-BOC更稳定,不易热分解,因此可以提高前烘温度(烘烤温度在聚合物玻璃化转变温度之上,热分解温度以下),减少胶膜内自由体积,避免环境中的氨进入胶膜内,减弱T-top现象[19,20]。叔丁基脱保护后转变成羧基,溶解速率快于酚羟基,利于提高光刻胶的对比度。
随着光刻胶及其聚合物的发展,诞生了一系列的酸不稳定酯基结构,如叔戊基、1-甲基环戊基、1-乙基环戊基、二甲基苄基和二甲基环丙基[21]。由于苯环结构在193 nm吸收较强,所以193 nm光刻胶树脂中多采用多元环酸不稳定结构,可以提高光刻胶的感度和耐蚀刻性,如图6所示金刚烷基[22]、1-乙基环己基[23]、降冰片类取代基[24]、倍半萜内酯[25]等,这些基团可以降低酸解过程中的排气量(outgassing amounts),保护昂贵的曝光机镜头不被污染。不同保护基团的酸解稳定性(acidolytic stability)、耐热性、亲疏水性直接影响光刻胶的性能,因此需要选择不同的单体结构以平衡光刻胶的性能。
(3) 缩醛缩酮:跟t-BOC和酯类保护基相比,缩醛缩酮属于低活化能保护基,在较低温度甚至室温就可以发生脱保护反应,环境中的碱还未进入胶膜内,脱保护反应已经完成,是解决T-top现象的一个有效方法[28]。缩酮在曝光过程中发生脱保护,因此比缩醛体系具有更好的环境稳定性,但是缩酮体系极易水解,影响合成和储存稳定性[29],目前,低活化能保护基以缩醛为主。烷氧基乙基结构中的烷基结构和体积大小直接影响光刻胶的性能[30],如图7所示,当R为乙基、叔丁基时,脱保护会释放气体,污染光刻机镜头,导致膜收缩;离子刻蚀过程中释放气体会导致光刻胶的蚀刻速率加快,蚀刻比下降。苯乙基或萘基乙基结构等可以降低挥发组分的含量,改善上述缺陷[28]。

图5 酸催化酯和醚脱保护
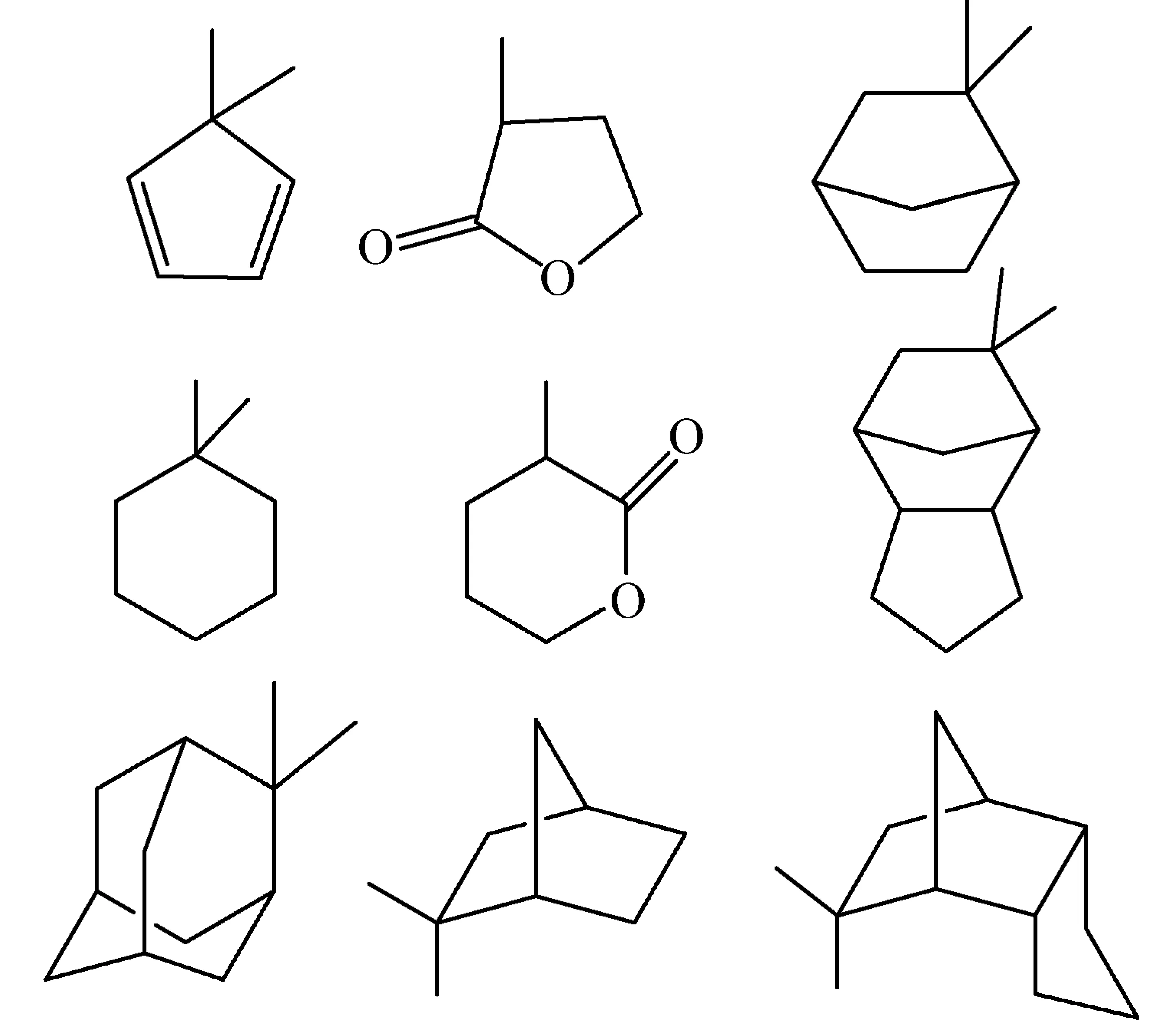
图6 部分环状保护基结构[26,27]
另外,缩醛结构还可作为羧基的保护基,Shirai等研究了金刚烷氧基甲基丙烯酸甲酯(admantyloxy methyl methacrylate)及其衍生物对光刻胶性能的影响[31];Wang等用丙烯海松酸(acrylpimaric acid)和1,3-金刚烷二羧酸与二乙烯基醚化合物反应,合成了一系列酯缩醛为保护基的聚合物[32,33],如图8所示。最近,Ober等[34]将缩醛结构引入到聚合物主链上,研究了聚(芳基缩醛)聚合物在EUV光刻中的应用,该聚合物经酸催化分子链断裂,分解成酚类等小分子。
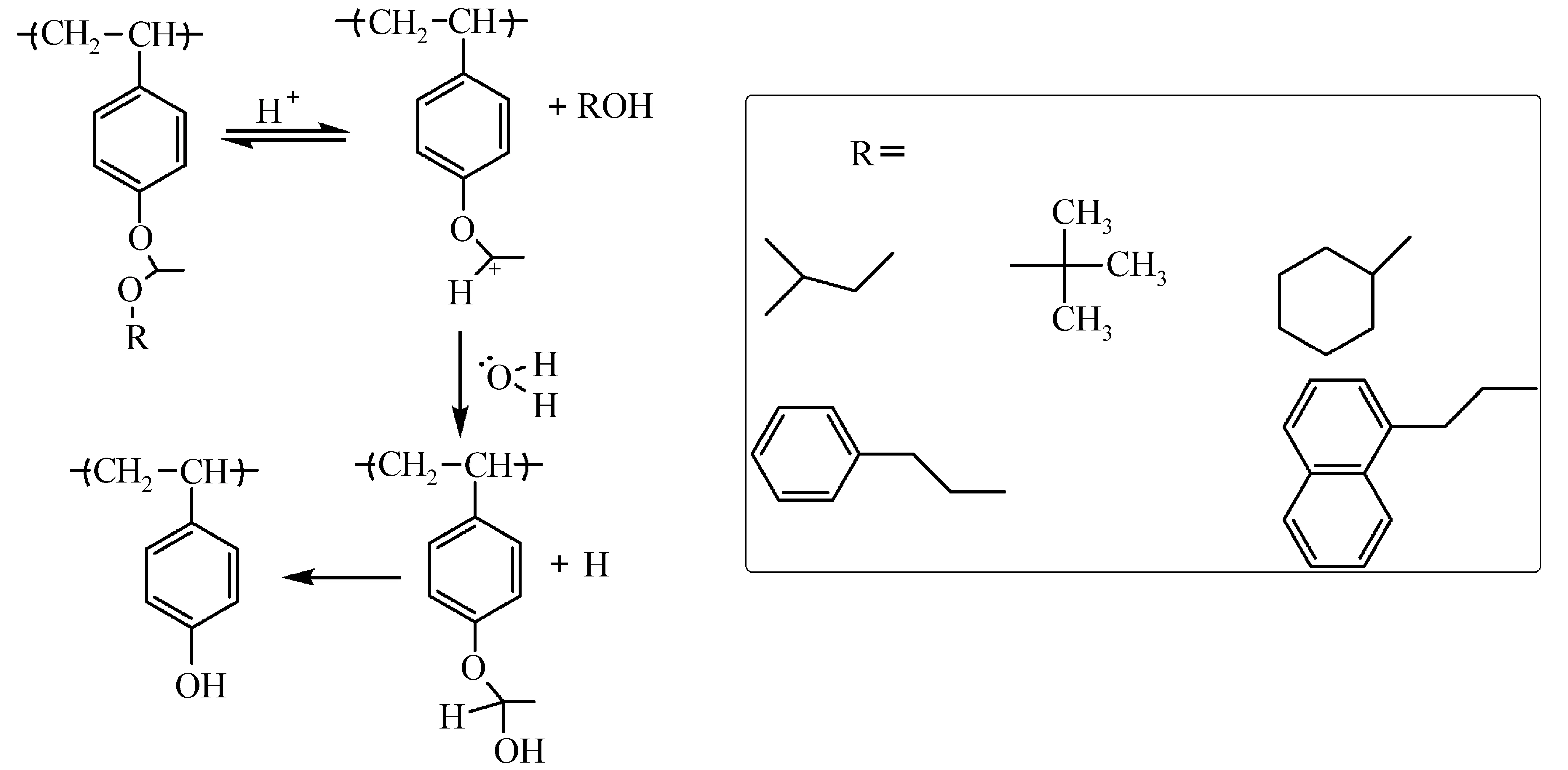
图7 缩醛脱保护反应机理[30]
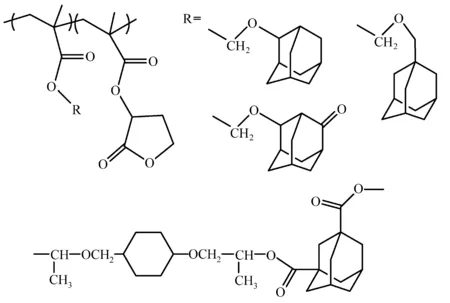
图8 甲基缩醛保护的聚合物结构[31,33]
(4) 开环反应: 含有叔酯结构的内酯和环状碳酸酯在热和酸作用下发生开环反应,产生的烯烃结构留在聚合物的骨架上,避免了挥发组分的产生[35]。如图9中聚(4-亚甲基-4H-1,3-苯并二恶英-2-酮)(poly(4-methylene-4H-1,3-benzodioxin-2-one) ),酸解后只产生CO2;四元环己内酯取代的苯乙烯结构在酸催化下开环,生成烯烃和羧基,无小分子生成[36]。
1.2 重排反应
重排反应同样可以改变聚合物的极性和溶解性,因此,可以利用酸催化重排反应制备化学增幅型光刻胶。
(1) 极性转变: 聚(4-乙烯基苯甲酸二甲基环丙基甲醇酯)(poly(2-cyclopropyl-2-propyl 4-vinylbenzoate,PCPPVB),热脱保护温度约为160 ℃,结构如图10所示。但是研究发现PCPPVB在160 ℃条件下加热,并未全部生成聚(4-乙烯基苯甲酸)和2-环丙基丙烯,其中10%的二甲基环丙基叔碳阳离子通过重排反应转化成4-甲基-3-戊烯基伯碳阳离子,进而与聚(4-乙烯基苯甲酸)反应生成聚(4-乙烯基苯甲酸-2-甲基-2-戊烯醇酯),在酸的催化下,重排反应比例可达66%。PCPPVB基光刻胶(PCPPVB与三苯基硫六氟锑酸盐)在100~130 ℃下烘烤,曝光区域脱保护成4-乙烯基苯甲酸极性单元,使用非极性苯甲醚显影,得到负性光刻胶图形(有机溶剂显影)。当PEB温度为160 ℃时,未曝光区域脱酯化反应率达到90%,存在大量的羧基,可溶于碱性显影液,曝光区域由于重排反应,仅含有33%的羧基单元,不足以溶于碱性显影液,该机理适用于水性显影负性光刻胶[37]。
上表中可以清晰地看到,教材内容由比较零散到相对集中;形式由摆一摆的具体形象到圈一圈的更为抽象;策略由阶段性的过渡到系统性的逐步提升。倍的教学后移,同时集中教学关于“倍”的数学问题,教学更富逻辑性与结构性。
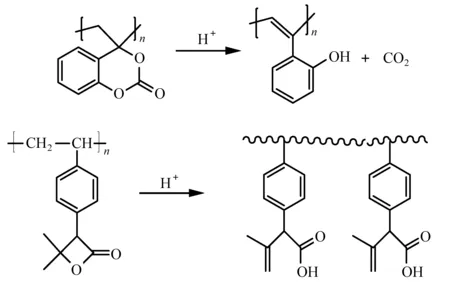
图9 Mass persistent光刻胶聚合物[35,36]
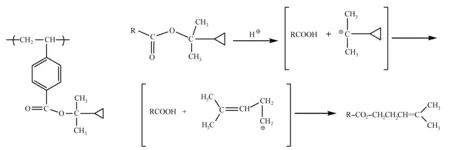
图10 PCPPVB的结构和酸催化重排反应[37]
聚甲基丙烯酸叔丁酯(poly(tert-butyl methacrylate),PTBMA)经曝光、后烘(post-exposre bake,PEB,低于150 ℃)、显影(TMAH水溶液)得到正性光刻胶图形。PEB温度为180 ℃时,曝光区域形成酸酐,而未曝光区域仍是PTBMA;再对整个胶膜进行过曝光,原来未曝光的区域发生脱保护反应,变成极性的羧基侧链,而原曝光区域是非极性的酸酐,碱性水溶液显影后得到负性图形[38]。因此可改变PEB温度以实现图形反转(image reversal)。
(2) 克莱森重排:当PHOST的羟基氢原子被环己烯氧基取代后,酸催化脱保护得到PHOST、1,3-己二烯和重排产物。1,3-己二烯通过克莱森重排反应在酚羟基的邻位进行取代,聚(4-苯氧基甲基苯乙烯)具有类似的重排反应[39]。克莱森重排反应使聚合物从非极性转变为极性,见图11。
(3) 频哪醇重排:频哪醇-频哪酮重排也是改变聚合物极性的有效方法[40]。将酚醛树脂、频哪醇化合物、PAG(二苯基碘鎓三氟甲磺酸盐)混合制备三组分化学增幅型光刻胶,在未曝光区域,频哪醇由于其亲水性,作为溶解促进剂;曝光区域,频哪醇重排生成酮或醛,起到溶解抑制的作用[40,41]。频哪醇化合物的结构直接影响光刻胶的性能。聚[3-甲基-2-(4-乙烯基苯基)-2,3-丁二醇]制备的化学增幅型光刻胶,极性醇(如异丙醇)显影时,未曝光区域的极性二醇聚合物溶解,曝光区域中由于酮结构极性弱,不溶于醇显影液,得到负性光刻胶图形,如图12所示[42]。对羟基苯乙烯和3-甲基-2-(4-乙烯基苯基)-2,3-丁二醇单元构成的聚合物,曝光部分频哪醇重排,不溶于TMAH水性显影液,未曝光部分羟基溶于水性显影液,得到水性显影的负性光刻胶[43]。Cho等[44]合成5-(2,3-二羟基-2,3-二甲基丁基二环[2.2.1]庚烯,将频哪醇重排反应应用于193 nm水性显影负性光刻胶(图13)。

图11 克莱森重排极性转变[39]
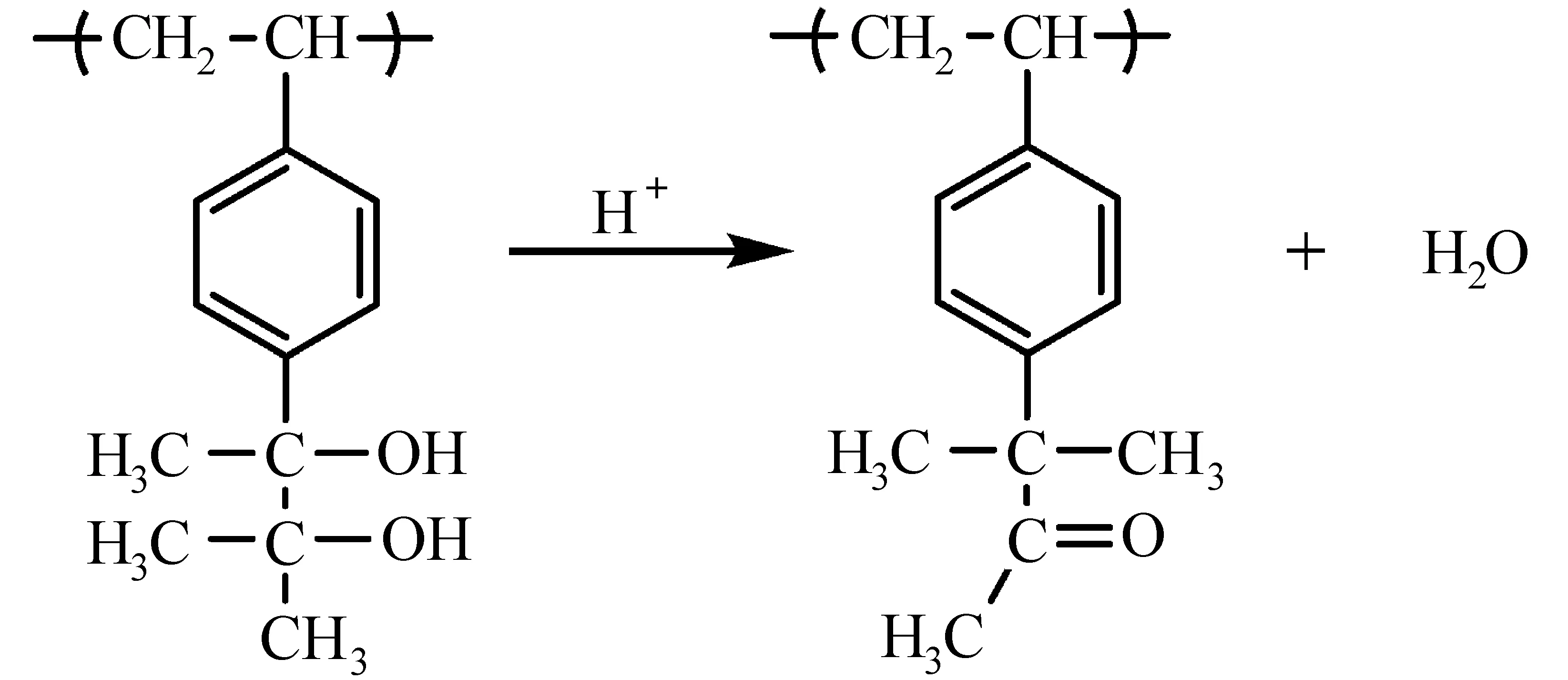
图12 频哪醇重排反应[40]
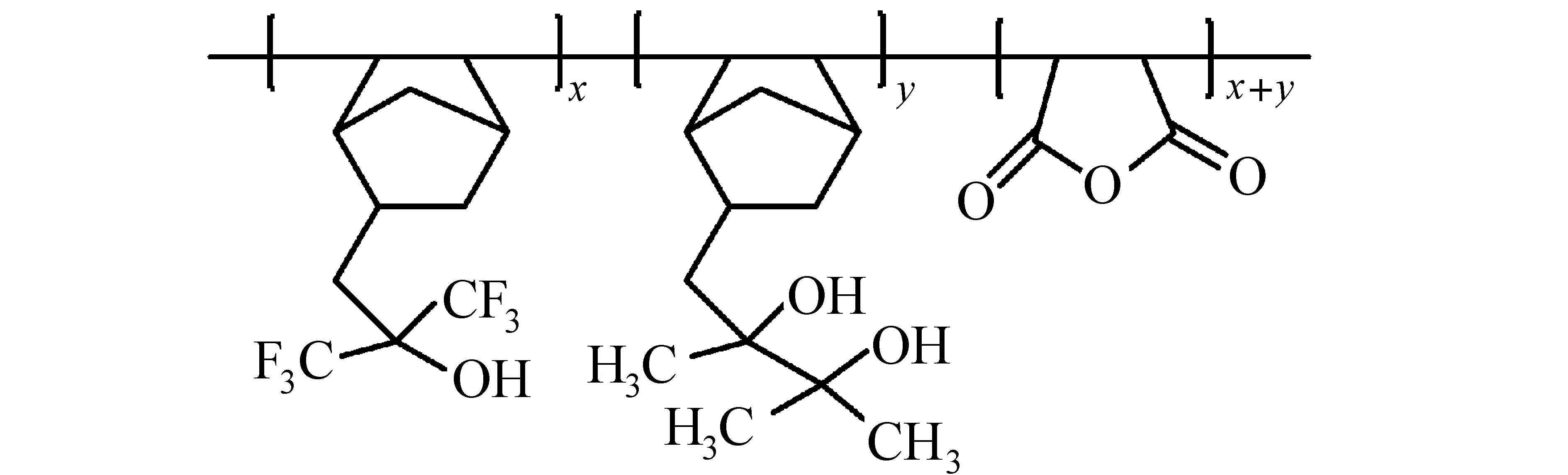
图13 频哪醇重排反应(193 nm负性光刻胶树脂)[44]
1.3 分子内脱水
聚[4-(2-羟基-2-丙基)苯乙烯]经酸催化脱水,产生稳定的叔苄基碳阳离子,经β-质子消除反应,形成侧链烯烃结构。酸催化分子内脱水反应如图14所示。分子内脱水反应将亲水性的醇转化为亲脂性烯烃,极性醇类(异丙醇,IPA)作显影液,得到负性光刻胶图形。但是,α-甲基苯乙烯部分经酸催化,会形成二聚体等交联产物(图15),因此用非极性有机溶剂显影,曝光部分不能全部溶解,无法得到光刻胶正性图像[45]。而聚[4-(1-苯基-1-羟基乙基)苯乙烯]由于苯环的位阻效应,曝光部分未发生交联反应,可以用非极性有机溶剂显影,得到光刻胶正性图像[43]。
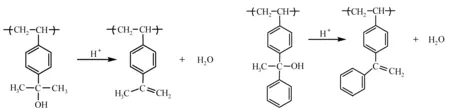
图14 酸催化分子内脱水[45]
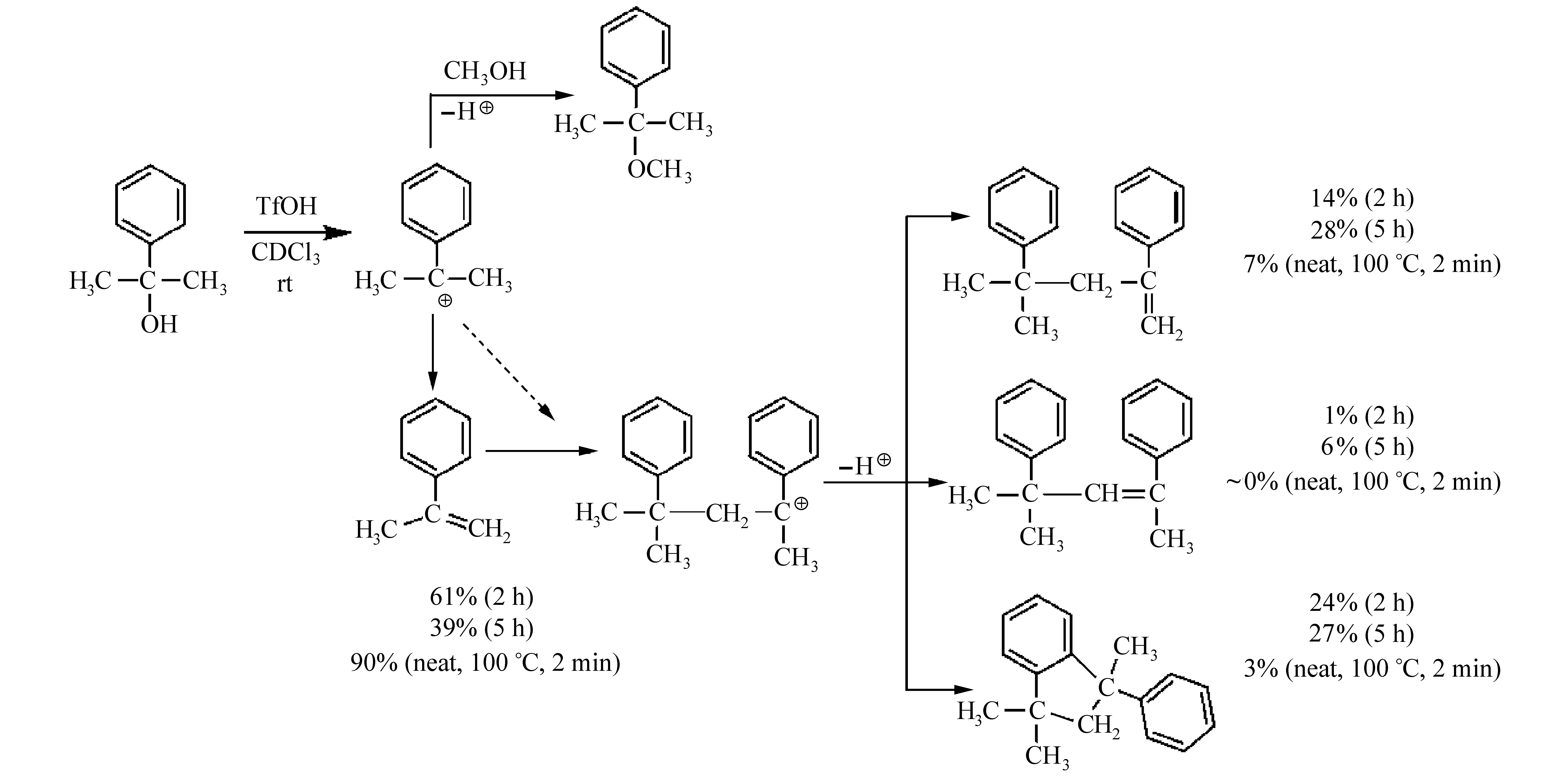
图15 酸催化二甲基苯基甲醇脱水[43]
1.4 缩合和酯化
酸催化缩合反应是主流的水性显影(TMAH水溶液)负性光刻胶体系。该类光刻胶主要由三部分组成:碱溶性树脂,交联剂(带有酸敏性亲电基团),PAG。酚醛树脂与六羟甲基三聚氰胺六甲醚(hexamethoxymethylmelamine,HMMM)在酸催化作用下形成交联网状结构(图16A)。PHOST替代酚醛树脂,用于DUV光刻[46]。PHOST与HMMM的反应机理不同于酚醛树脂,而是酚羟基与HMMM发生交联反应,导致聚合物分子量增大,碱溶性酚羟基减少(图16B)[47,48]。PEB的温度和时间、PAG和交联剂的结构直接影响交联程度,对光刻胶的性能(感度、对比度、分辨率)起着重要作用。
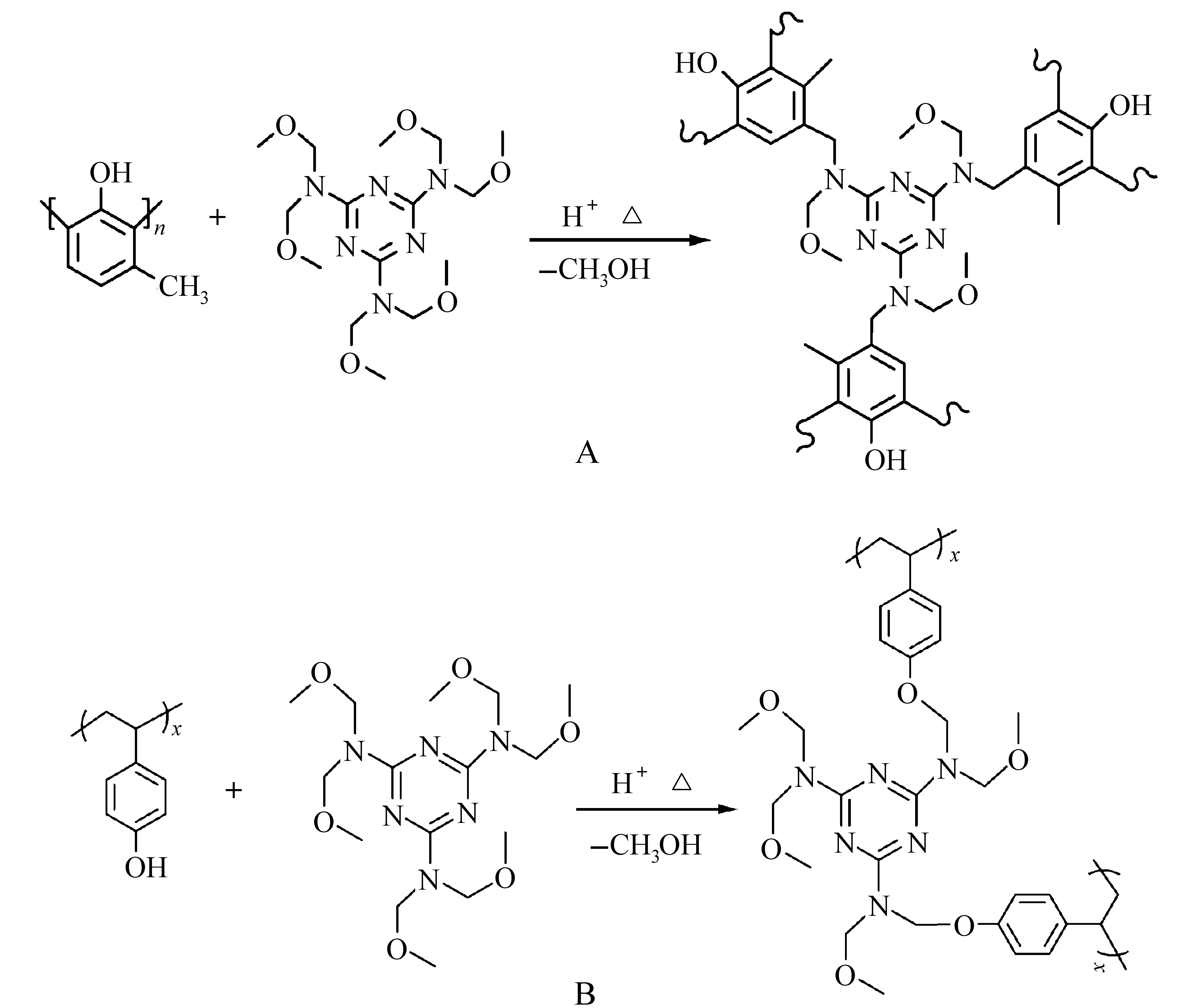
图16 酚醛树脂和PHOST与HMMM反应机理[46]
乙酸苄酯衍生物作为一种亲电子试剂,在酸作用下产生稳定的苄基碳阳离子,释放乙酸;碳阳离子在苯环上发生亲电取代反应,形成交联网状结构,如图17所示[49]。
当体积庞大的取代基与苯环发生亲电取代反应后,即使没有交联反应,也可以显著降低树脂在碱溶液中的溶解速率。单官能团的亲电试剂N-羟基和N-乙酰氧基甲基酰亚胺可与PHOST进行酸催化缩合,生成C—烷基和O—烷基产物,降低曝光区域的溶解速率[50],如图18所示。酸催化苯酚和醛反应是合成酚醛树脂的重要方法之一,该缩合反应机理可用于制备负性光刻胶。Ito等[50]将PHOST、醛和PAG共混,醛作为亲电试剂在酸催化下与PHOST发生取代反应,得到羟甲基化PHOST,随后继续与PHOST发生取代反应,如图19所示。PHOST与醛加成-缩合反应增加了聚合物的分子量,改变了树脂的溶解速率。
上述光刻胶树脂本身需要具有反应位点,需要有交联剂或亲电试剂参与反应。呋喃环对亲电试剂具有高反应性,而呋喃甲醇在酸催化下容易产生稳定的碳阳离子(亲电试剂),因此,呋喃甲醇衍生物容易发生亲电取代自缩合反应,不需要交联剂[49],如图20所示。
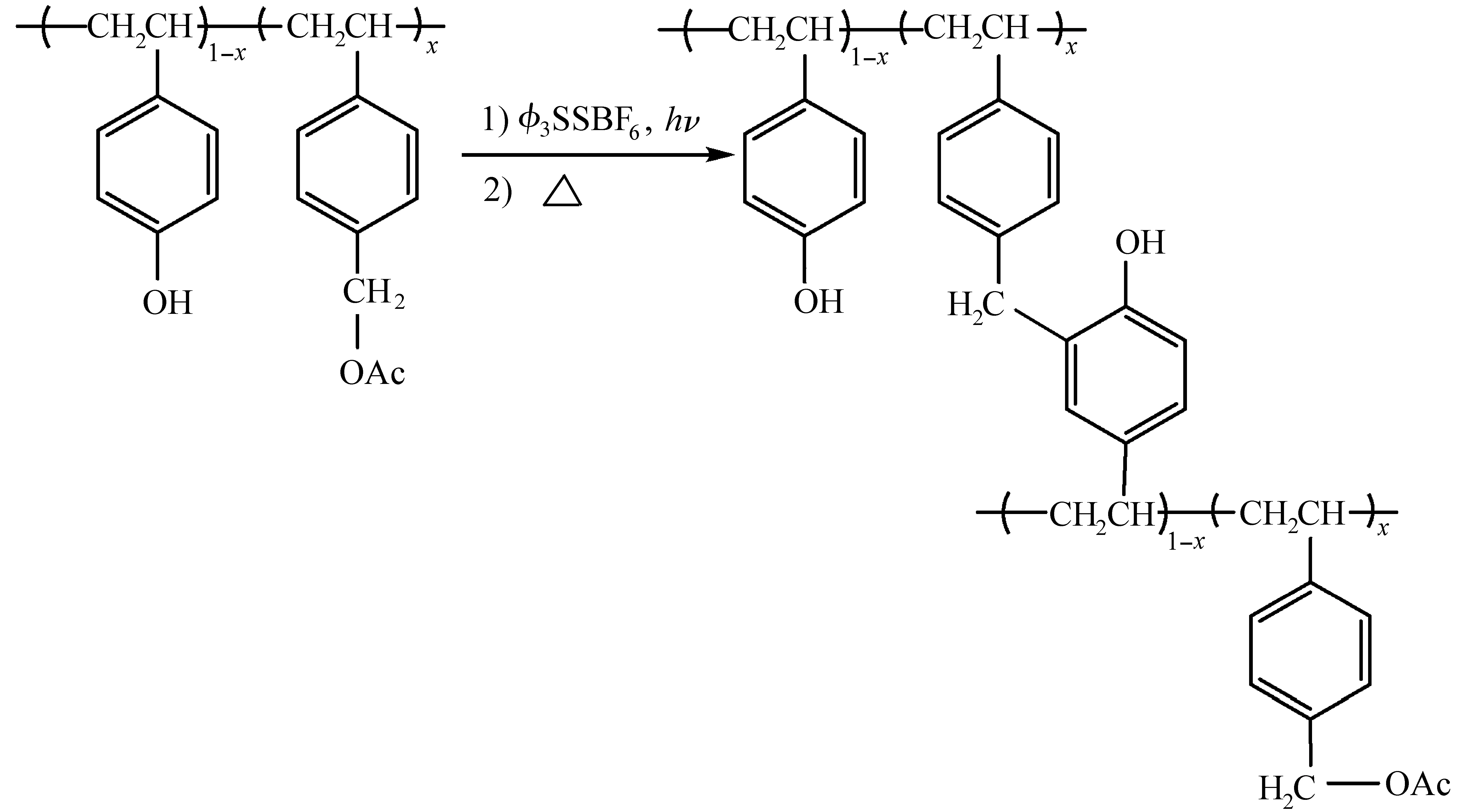
图17 亲电芳环取代反应[49]
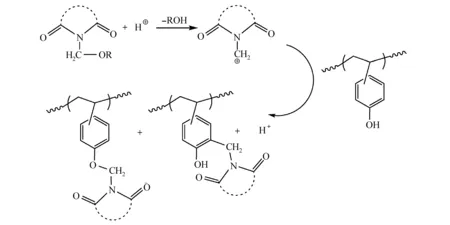
图18 N-乙酰氧基甲基酰亚胺与PHOST的C、O—烷基化反应[50]
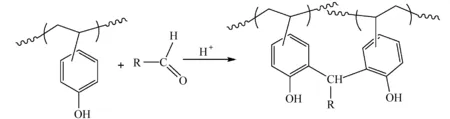
图19 酚和醛缩合反应[50]
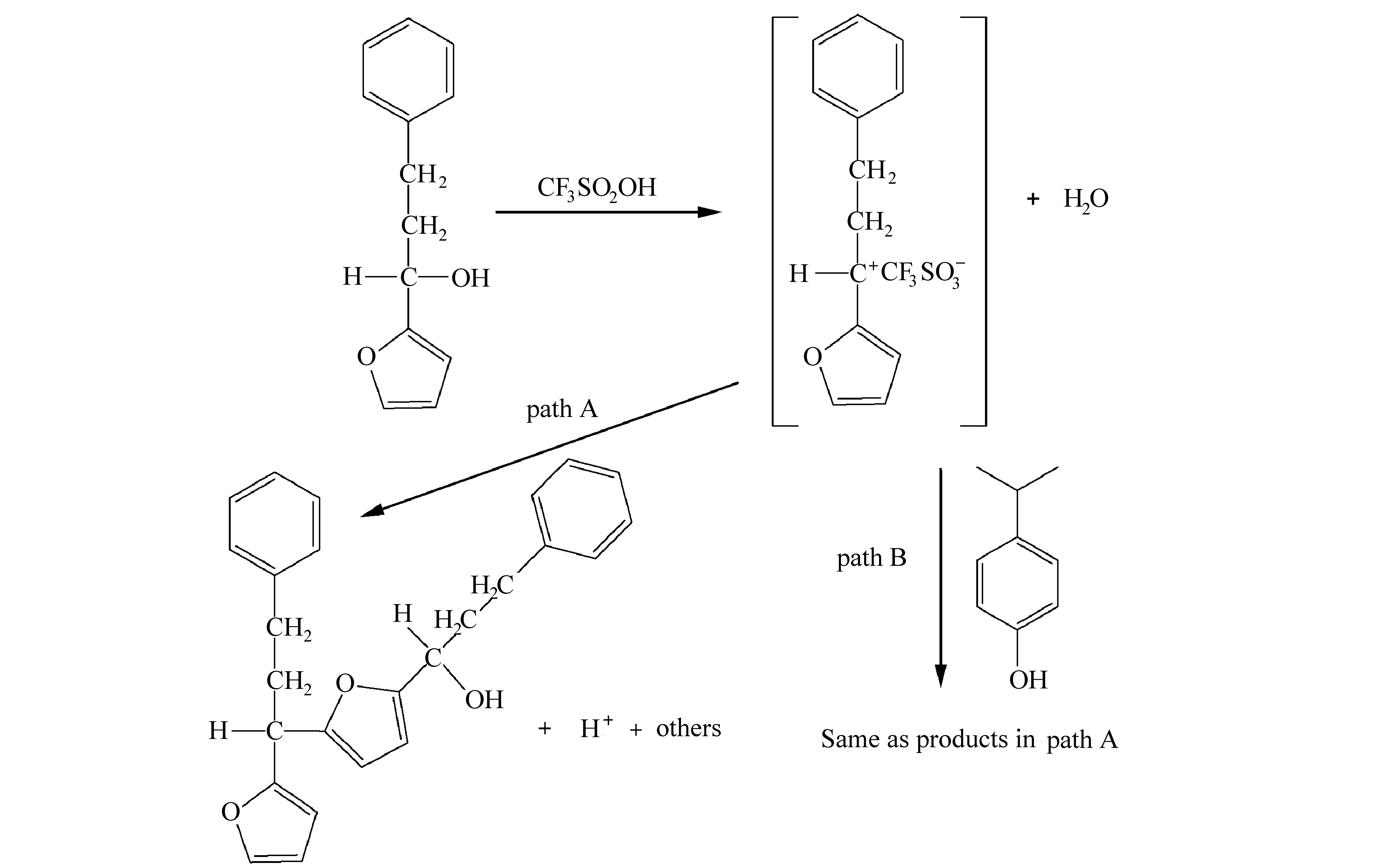
图20 酸催化聚合呋喃甲醇自缩合反应[49]
硅烷醇化合物被用于水性显影负性光刻胶。Shiraishi等[52]用酚醛树脂、硅烷醇化合物和PAG制备光刻胶,曝光区域的硅烷醇发生缩合反应生成聚硅氧烷结构(图22),疏水性的聚硅氧烷排布在酚羟基周围,阻挡显影液扩散,有效降低溶解速率,起到了溶剂抑制的作用,未曝光区域亲水的硅烷醇起到溶解促进的作用[53,54]。
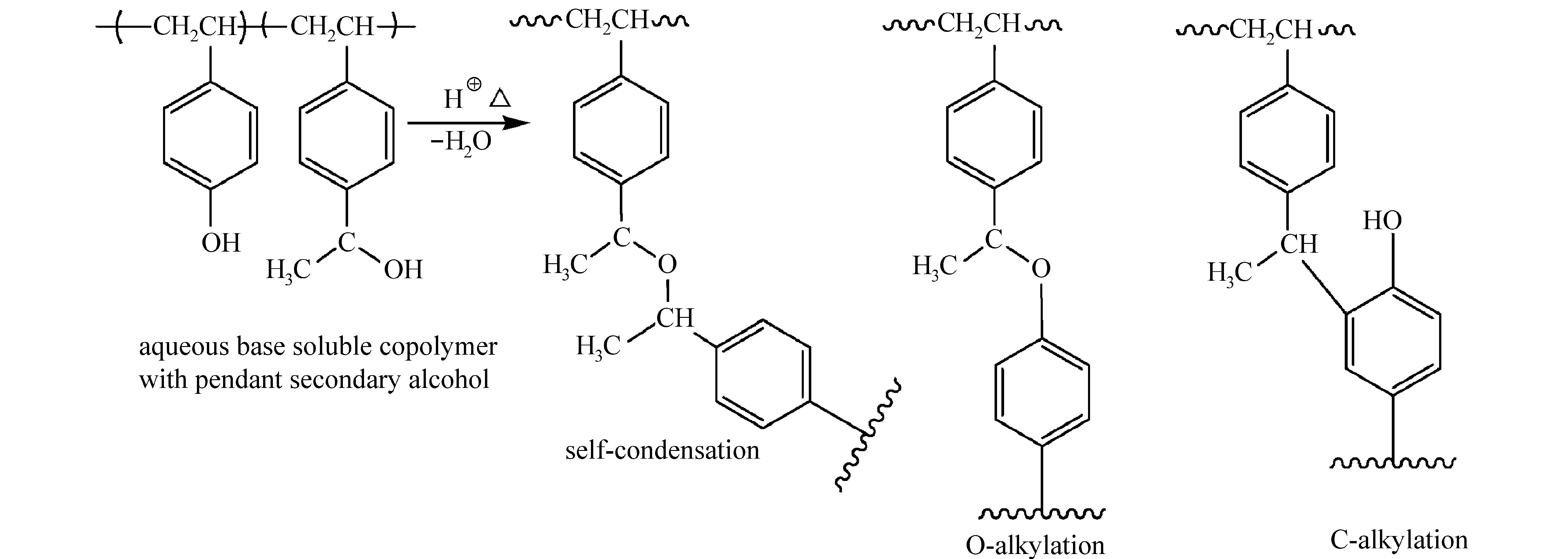
图21 酸催化分子间脱水自缩合反应和C、O烷基化反应[51]
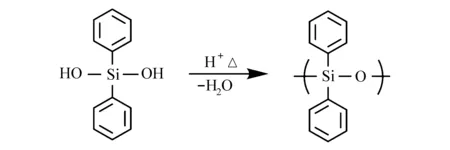
图22 硅烷醇缩合反应[53]
苯环在193 nm处有吸收,因此ArF光刻胶树脂多采用脂肪环结构,所以不存在苯环亲电取代反应。ArF负性光刻胶主要是通过曝光部分交联反应降低溶解速率。Iwasa等[55,56]合成了一系列ArF光刻胶聚合物,发现在聚合物中引入极性羟基、羧基、环氧基和内酯基可以提高胶膜与基材之间的粘附力;羧基和羟基的含量可以调节聚合物的溶解速率;羟基与交联剂(1,3,4,6-四(甲氧基甲基)甘脲、HMMM等)反应生成网状结构,不溶于碱性显影液,见图23。
此外,Lee等[57]研究了甲基丙烯酸羟乙酯结构中的羟基酯化反应(图24)。
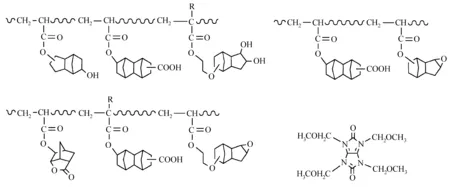
图23 基于羟基缩聚反应的ArF光刻胶树脂结构[55,56]
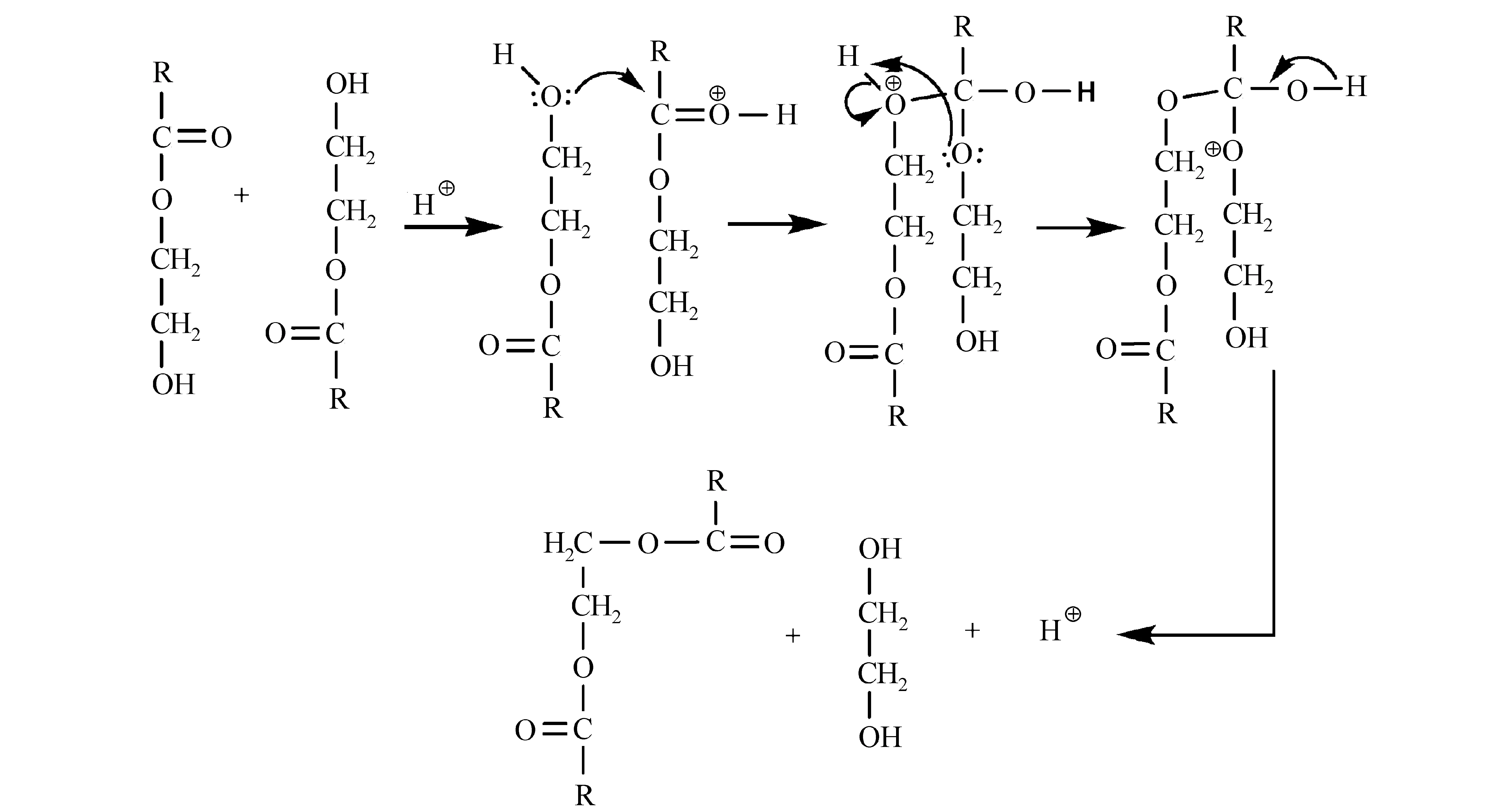
图24 基于2-羟乙基酯交换的甲基丙烯酸酯聚合物负性光刻胶[57]
Hattori等[58]合成了含有γ-羟基酸(γ- hydroxy acid)结构的脂环族聚合物,γ-羟基酸在酸催化下发生酯化反映生成内酯,但γ-羟基酸的内酯化反应对聚合物的结构有选择性(图25)。同时,Yokoyama等[59]研究了δ-羟基酸(δ-Hydroxy Acid)内酯化反应在负性光刻胶的应用。
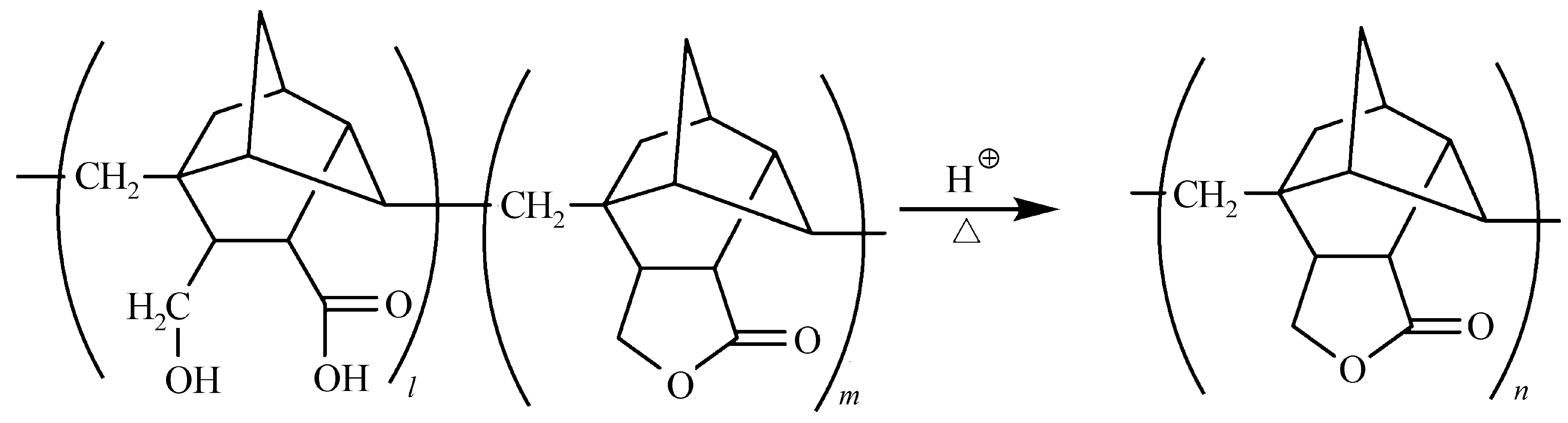
图25 酸催化脂环族聚合物中γ-羟基酸的反应[58]
1.5 交联聚合
SU-8胶是典型的交联聚合反应机理光刻胶,在酸的催化作用下环氧开环聚合。SU-8胶主要应用在MEMS领域,用来制备高深宽比微结构与微零件[60]。此外,有文献报道了一系列基于环氧、环硫聚合物的负性光刻胶[61,62]。由于该类负性光刻胶用有机溶剂显影,存在溶胀效应,易导致图形尺寸变形,影响其在高分辨率领域的应用。
环氧单体与碱溶性树脂(PHOST,酚醛树脂)共混[63]、环氧单体与碱溶性单体共聚合成碱溶性环氧树脂[64],可制备碱溶性负性光刻胶,降低光刻胶的溶胀效应。采用含有环氧基团的分子玻璃作为成膜树脂,可以有效改善光刻胶的性能。由于分子玻璃具备成膜性好、单一分子量分布等特殊性能,制备的光刻胶在电子束光刻条件下可达到高分辨率[65]。图26分别是阳离子开环聚合所用的SU8树脂和分子玻璃的结构式。
由于环氧开环聚合是阳离子聚合,即使酸被碱性添加剂中和掉,活性链还是会继续引发聚合反应,造成未曝光部分发生交联反应,因此需要引入亲核试剂作为链终止剂。而鎓盐(如三苯基硫鎓三氟甲基磺酸盐)在曝光区域可以产生酸,引发环氧聚合,在未曝光区则可以作为链终止剂,终止扩散到未曝光区域的活性链(图27)。因此在环氧交联聚合光刻胶中可以选用三苯基硫三氟甲烷磺酸盐作为PAG[5]。
1.6 解聚反应
聚甲基丙烯酸甲酯(polymethylmethacrylate,PMMA)是第一款解聚型光刻胶树脂,在电子束和DUV光刻中,聚合物主链断裂分解成低分子量碎片,可增强溶解性[66,67]。虽然它表现出优异的分辨能力,但是主链断裂需要的能量高,而且PMMA脂肪链结构的耐蚀刻性较差。
醛通过阴离子或阳离子聚合生成聚醛,但是聚醛与单体存在可逆平衡,在室温下会发生解聚反应,因此可以通过控制反应方向实现聚合物跟单体之间的转化,改变溶解性。对聚醛进行烷基化或酰化封端,可以提高聚合物的热稳定性,抑制解聚反应;在聚醛中引入芳环结构可以改变醛的结晶性,得到非晶无定型结构的醛,提高聚合物在有机溶剂中的溶解性[68],涂布后得到各向同性的光刻胶胶膜。缩醛封端的聚邻苯二甲醛(polyphthalaldehyde,PPA)在光酸作用下缩醛键断裂,然后发生解聚反应,重新分解成单体,见图28。该聚合物在电子束作用下会迅速分解挥发[68]。因此将该聚合物用电子束曝光时,曝光部分直接分解挥发,不需要后烘显影工序就可以得到图形,称为自显影电子束光刻胶(self developing electron-beam resist)[69]。溴和氯取代的聚苯甲醛在低能量曝光后,不会发生自显影,而需要在后烘过程中(>100 ℃)完成分解反应,可以避免挥发组分对镜头的污染[70]。为了改善PPA的耐蚀刻性,Ito等[71]将聚邻苯二甲醛充当做溶解抑制剂,与PAG一起加入到酚醛树脂中制备三组分正性光刻胶,也可以引入硅元素提高此类光刻胶的耐氧离子蚀刻性[72]。
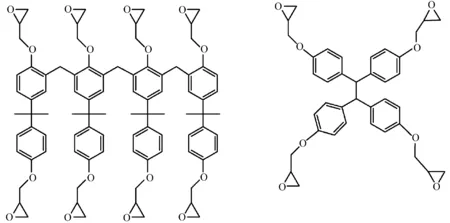
图26 用于阳离子开环聚合交联的环氧树脂[60,65]
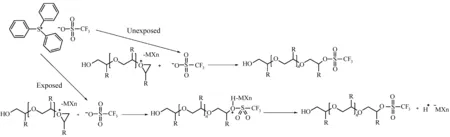
图27 “光转换”亲核试剂(PSNs)控制聚合反应[5]
聚α-乙酰氧基苯乙烯(poly a-acetoxystyrene,PACOST)高温(220 ℃)分解成聚(苯乙炔)和乙酸。酸催化可以降低聚合物主链的脱酯化温度。因此在PACOST中加入PAG,曝光部分在PEB过程中会发生解聚反应(图29),极性和分子量发生变化,用二甲苯显影可得到正性图形。 PACOST的刚性环结构提高了光刻胶的耐热性(Tg> 200 ℃),但在显影过程中易裂[73]。此类解聚反应是从聚合物链末端开始的,因此要考虑聚合物的末端结构对解聚反应、光刻胶性能的影响,所以不同结构的引发剂、不同的聚合方法得到的聚合物稳定性也不同[74]。
含叔碳原子的聚碳酸酯在200 ℃裂解成二醇、烯烃和二氧化碳[75,76]。同样,在酸的催化作用下,聚合物裂解温度降低,C—O键断裂,主链发生解聚反应。二醇(1,4-二羟基-1,2,3,4-四氢化萘或1,4-二羟基-2-环己烯)与二溴甲烷通过相转移缩合制备的聚合物在150 ℃不分解,但在酸催化下80 ℃便可裂解,得到芳族产物(苯或萘)和甲醛水合物。该类聚合物深紫外吸吸光度很低,因此可掺入PAG制备化学增幅性光刻胶[77]。二氯嘧啶与二醇制备的聚醚,主链中含有二烷氧基嘧啶单元,经酸催化裂解成嘧啶酮和二烯[78]。该类主链断裂聚合物结构如图30所示。
2 结论
本文根据化学增幅型光刻胶中聚合物的反应机理进行分类阐述,其中脱保护、重排反应、分子内脱水改变了聚合的极性,适用于正性或负性(NTD)光刻胶。解聚反应式使得聚合物的分子量减低,适用于正胶;分子间脱水、缩聚酯化和聚合造成交联反应,聚合物的分子量增大,形成交联网状结构,不溶于有机溶剂,更适用于负胶。
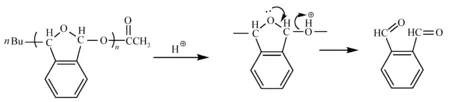
图28 酸催化聚苯二醛解聚反应[68]
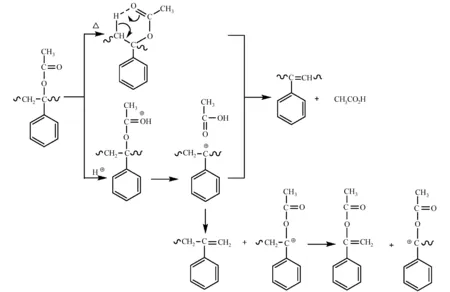
图29 PACOST的热分解和酸催化分解机理[73]

图30 酸催化主链断裂[76-78]
目前,化学增幅机理已在I线光刻胶、DUV(248 nm、193 nm干法和浸没式)光刻胶和EUV光刻胶中得到广泛应用。其中,脱保护反应占据重要地位。高活化能KrF光刻胶如TOK(东京应化)的TDUR-P534;中等活化能KrF光刻胶种如美国Dow UV1610; 低活化能KrF如日本信越开发的SEPR302、SEPR555,住友化学(Sumitomo Chemical)的PEK系列,TOK的DP-3262等;低活化能类型的ArF光刻胶如信越X系列的X192/X193/X212等。在实际应用中,需要根据光刻图形种类(线(line)、孔(hole)、槽(trench))、膜厚、感度等因素选择合适的光刻胶和聚合物。
猜你喜欢
新材料产业(2021年5期)2021-10-29 01:09:11
大学化学(2021年7期)2021-08-29 12:21:30
液晶与显示(2021年2期)2021-03-02 13:38:40
新材料产业(2019年10期)2019-12-23 08:28:12
通信技术(2019年8期)2019-09-03 08:57:08
时代英语·高一(2019年5期)2019-09-03 02:09:34
国际呼吸杂志(2019年4期)2019-03-12 01:08:08
股市动态分析(2017年41期)2017-11-01 13:16:12
电测与仪表(2016年11期)2016-04-11 12:20:42
电源技术(2015年5期)2015-08-22 11:18:28
