
IRF5与SLE的关系及在免疫细胞中的作用研究进展
2020-05-25 10:28李文英廖湘平
中南医学科学杂志 2020年3期
李文英, 廖湘平
(1.南华大学,湖南 衡阳 421001; 2.南华大学附属郴州医院肾内科,湖南 郴州 423000)
系统性红斑狼疮(Systemic lupus erythematosus,SLE)是一种慢性特异性自身免疫性疾病,其主要特点是多种致病性自身抗体形成、补体激活及免疫复合物沉积。自身抗体和免疫细胞攻击正常组织,导致包括肾脏、皮肤、关节、中枢神经系统和心血管系统在内的多种器官的损伤。SLE血清中存在大量以抗核抗体为主的自身抗体,其形成的免疫复合物中的核酸可被细胞Fcγ受体结合后运送至内体Toll受体(Toll-like receptors,TLR),这种异常递送可能激活异常的抗病毒免疫,通过联级放大作用,产生大量IFN-α,致使炎症加重[1]。在上述过程中,IRF5发挥了重要作用。IRF5是参与调节先天性免疫应答、免疫细胞发育和肿瘤发生的IRF家族的成员之一(共有10个成员IRF1-IRF9及vIRF)。大量研究表明IRF5多态性与一系列人类自身免疫性疾病(包括系统性红斑狼疮、硬皮病,类风湿性关节炎,溃疡性结肠炎)的发病风险有关。近年来,IRF5在调控神经病理性疼痛,心血管疾病,同种异体排斥反应和代谢功能障碍等方面的作用也受到了关注。大量研究表明SLE患者中IRF5表达异常,推测IRF5通过影响多种免疫细胞的活性和(或)功能控制免疫反应而使机体致病。本文将重点阐述IRF5与SLE的相关性及IRF5在树突状细胞、淋巴细胞、单核细胞-巨噬细胞等多种免疫细胞中的作用。
1 IRF5和TLR信号通路
IRF5是IRF家族的成员之一,主要通过TLR信号途径参与免疫应答(图1),常见的TLR是位于胞质的TLR7、TLR8和TLR9。TLR7、TLR8识别ssDNA细菌和ssRNA病毒,TLR9识别dsDNA病毒或细菌中的DNACpG基序,这些配体与系统性红斑狼疮中的抗核抗体及含有这些抗体的免疫复合物的识别相关。据报道,来自SLE患者外周血单个核细胞中TLR7、TLR9的mRNA表达上调,且其水平与IFN-a的表达相关。识别上述配体后,通过信号中间体MyD88激活IRF5并启动信号传导。作为具有Toll/白介素1结构域的衔接蛋白,MyD88募集了白介素1受体相关激酶(IRAK-4)[2]。IRAK-4结合并磷酸化IRAK-1或IRAK-2,后者又募集肿瘤坏死因子受体相关因子6(TRAF6)。TRAF6是E3泛素连接酶,与IFR5相互作用并将K63-Ub链添加至IRF5使其泛素化。以上这些事件为IRF5核转位奠定了基础,但这种泛素化作用仍不够,所以还需要IκB激酶β(IKKβ)磷酸化调节IRF5蛋白表达和活性。相比IKKβ诱导IRF5磷酸化促进IRF5的活化,IKKa诱导IRF5磷酸化可能抑制TRAF6介导的IRF5泛素化,从而起反作用。活化的IRF5经历同型二聚体形成和核转位,然后与编码IFN-a和其他促炎因子的基因的特定序列结合,从而诱导这些基因的转录[3]。更高的IRF5活性可以诱导丰富的IFN-a和促炎因子,这可能促使狼疮疾病的发生与发展。
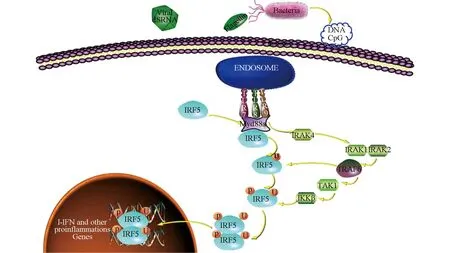
图1 IRF5参与免疫应答的TLR信号途径
2 IRF5基因结构
IRF5的激活不仅受不同酶的调控,其基因表达也很复杂。IRF5基因染色体7q32上,由50个非翻译区(UTR)中的8个编码外显子和4个非编码第一外显子(1a,1b,1c,1d)组成。外显子1c启动子含有干扰素刺激响应元件(ISRE)通过形成STAT1/2-IRF9复合物(干扰素刺激的基因因子3,ISGF3)对I型IFN响应[4]。4个非编码第一外显子启动子均位于外显子2中的ATG翻译起始密码子,均有3个转录起始位点,因此,经不同剪接可产生9个IRF5转录异构体(v1-v9),已经证明不同转录异构体在不同细胞类型中差异性表达。选择性剪接产生的大量新型IRF5同种型在SLE患者中特异性表达,意味着其可以作为自身免疫性疾病的生物标志物。具有不同细胞特异性表达、功能和活性的几种IRF5转录异构体也可因外显子6的插入/缺失发生选择性剪接而产生[5]。外显子6中30 bp框内缺失形成的indel-10具有启动IRF5靶基因转录的独特能力。外显子6中插入48 bp后称为剪接变体SV-16,这一突变与IRF5-v1和IRF5-v5的转录有关,且可促进细胞凋亡和细胞因子释放。然而,indel-10共表达可以中和SV-16的这些作用[5]。IRF5还具有一个CpG岛,包括转录因子Sp1的结合位点。活性CGI启动子通常富含Sp1结合位点,因为Sp1的DNA结合反过来可以将RNA聚合酶II和TATA结合蛋白吸引到该区域,即使在没有TATA盒的情况下也能促进转录[4]。CGI区域富含非甲基化GC,与转录起始相关,因而外显子1a的转录物高表达,所以CpG岛甲基化,可能导致IRF5基因沉默。
3 IRF5基因多态性与SLE
IRF5基因多态性一直是研究的热点,全基因组关联研究(GWAS)证实,IRF5风险单倍体与SLE易感性呈显著性相关。不同人群中SLE表型有所差异,这可能是IRF5单核苷酸多态性(SNP)在不同遗传背景下的表达结果。最初在欧洲血统中发现IRF5中的三种变异基因型(rs2004640、rs10954213和rs77571059)与SLE风险增加有关[6]。目前,研究发现IRF5 NSP中与SLE易感性关联最紧密的主要是rs2004640单倍体。最新的一项Meta分析[7]指出目前的研究表明携带rs2004640 t等位基因的个体与SLE的高风险相关,IRF5 rs2004640多态性与SLE易感性相关。
rs10954213多态性在SLE中也起着特殊的作用。对IRF5和SLE中rs10954213的多分形关系进行了11项研究,结果表明IRF5中rs10954213和TCA单倍型(rs2004640-rs2070197r-rs10954213)等位基因与不同种族的SLE患病风险增加有关。此外,rs10954213被证明对复发性妊娠损失(RPL)具有保护作用[8]。在克里特人群中也验证了IRF5可以通过IFN水平升高来调节SLE的风险[9]。Hammad等[10]在埃及儿童中首次研究了IRF5多态性研究,发现rs10954213,rs2004640和rs2280714、rs2004640 T等位基因及TT基因型和GTA单体型可作为埃及儿童SLE发生的危险因素,rs2004640 T等位基因的存在增加了埃及SLE儿童肾炎发展的风险。虽然GWAS已经检测到许多SLE易感基因,但在不同的种群中IRF5 SNP与SLE的遗传关联存在许多差异,继续研究SLE在多人群中的遗传基础仍是重要且有必要的。
4 IRF5在免疫细胞中的作用
4.1 树突状细胞中的作用
在病毒感染的早期反应中,大多数免疫细胞都能产生Ⅰ型干扰素,浆细胞样树突状细胞(pDC)是最主要的生产者。而且IRF5在pDC中高度表达并参与促炎细胞因子和趋化因子的产生,尤其是TNF和IL-6。Steinhagen[11]使用RNA干扰技术,明确IRF5和IRF8作为正面和负面调控因子在pDC产生干扰素和细胞因子的诱导机制中发挥了关键作用:TLR9 TLR激活的CAL-1 pDC细胞系(功能上类似于原代人类pDC)中,IRF-5在MyD88/TRAF6(泛素连接酶)依赖性诱导IFN-β和IL-6的诱导中发挥关键作用。CAL-1 pDC细胞系是研究pDC信号通路的重要工具,但Steinhagen的实验结果可能是因细胞恶性转化引起的供体特异性效应和异常,随后通过测序证实存在CAL-1细胞中的IRF8基因不含有突变[12]。与此同时,证明了IRF5在上调人类pDC的绝大多数基因中发挥关键作用,而且IRF8是通过与IRF5的直接相互作用起到制动作用。MyD88依赖性IRF5通过调节CCL19/CCL21来促进pDC向腹膜迁移。与野生型B6小鼠相比,用Pristane处理的IRF5-/小鼠的腹膜腔中pDC的数量显着减少,说明在缺乏IRF5的情况下MRL/lpr小鼠的炎性细胞因子和趋化因子的产生受到了抑制,干扰素水平有所下降,MRL/lpr小鼠类SLE症状也得到缓解。经IFNβ预处理的FL-DC(从在fms样酪氨酸激酶3配体中培养的BM细胞获得的DC)中的LPS诱导的IFNα合成依赖于TLR4、TRIF(Toll/IL-1受体含有结构域的衔接蛋白诱导IFNβ)和MyD88通路,强调IRF5参与Flt-3L DC产生的TLR9诱导的IFN-α和IFN-β产生。此外,来自DOCK2-/-小鼠的浆细胞样树突状细胞(pDC)对于TLR7和TLR9配体的应答产生IFN-α和IFN-β的能力受损,通过逆转录病毒转导的DOCK2的异位表达恢复了pDC的发育。因此,DOCK2基因突变可导致pDC产生Ⅰ型干扰素的能力下降和免疫应答减弱。
4.2 IRF5在巨噬细胞中的作用
IRF5是巨噬细胞极化的关键调节因子。除了源自骨髓中产生的单核细胞的巨噬细胞之外,不同来源的组织巨噬细胞也可能在炎症中发挥关键作用,有动物实验表明在显著激活的巨噬细胞和其它髓系细胞大量浸润到淋巴组织和肾脏中,启动和促进适应性免疫应答,从而导致SLE的发生[13]。巨噬细胞根据不同病原体的不同反应可以分为不同的亚型:M1和M2巨噬细胞,M1巨噬细胞是具有更强的侵袭性表型,可以介导有效的炎症反应,M2巨噬细胞介导较少的温和的炎症反应并促进细胞增殖和组织修复。SLE特征之一就是表现为不可控的炎症反应,M1巨噬细胞在疾病发病机制中起重要作用。不同的巨噬细胞亚型不仅以细胞因子释放的差异为特征,而且显示关键转录因子的差异表达。IRF5被证明是涉及促炎性M1巨噬细胞极化的主要调节因子[14],其表达也在巨噬细胞分化过程中被诱导。IRF5激活巨噬细胞诱导促炎性细胞因子的表达,如IL-6,IL-12b和IL-23a,同时抑制抗炎细胞因子如IL-10的转录,IRF5通过调控细胞因子谱影响M1巨噬细胞极化。IRF5作为M1巨噬细胞表型分化的主要调节剂并通过促进炎性细胞因子,趋化因子和共刺激分子的转录激活,导致效应性T细胞应答效率的提高。在急性胰腺炎(SAP)小鼠模型及糖尿病大鼠模型中也证实了IRF5在巨噬细胞极化中的关键作用。
4.3 IRF5在B淋巴细胞中的作用
IRF5在B细胞成熟、分化和抗体类别转换过程中发挥重要作用。GWAS已经鉴定了人类基因组中的IRF5和PRDM1等狼疮易感基因。疮样小鼠中,IRF5被TBK1和MyD88激活形成同源二聚体,激活的IRF5诱导PRDM1基因的转录生成B淋巴细胞诱导成熟蛋白-1(Blimp-1)[15]。Blimp-1是维持抗原特异性免疫反应的重要转录因子。在分子水平,IRF5通过调节Blimp-1的表达部分促进B细胞成熟,促进SLE的发生与发展。在小鼠中,IgG2a和IgGb亚类自身抗体被认为是致病性最强的,在人类中,鼠IgG2a和IgG2b同种型被认为分别对应于IgG1和IgG3。与其鼠类相似,IgG1和IgG3是SLE患者中最主要的自身抗体亚型。在不同生物体内,特定类型的抗体引起SLE的临床表现,如免疫复合物沉积引起的狼疮性肾炎和补体激活介导的炎症。IRF5可能影响SLE中浆细胞抗体类别转换,使IgG抗体分泌增多而其他类型如IgM减少。IRF-5-Ikaros轴是IgG2a/c类别转换的关键调节剂。在正常小鼠的B细胞发育中,IRF-8激活ikzf1启动子而促进Ikaro转录,继而抑制IgGa/c基因座,而依赖于MyD88表达的IRF-5抑制IRF-8介导的IKaro转录,间接控制了IgG向更易致病的IgG2a和IgG2b亚型类别转换[16]。此外,Ikaros通过促进自身反应性B细胞失能和抑制TLR信号,起到了保护机体自身免疫的作用,从而抑制狼疮的发生发展。
在小鼠红斑狼疮模型中,IRF5缺陷在疾病发病机制中具有重要作用。IRF5缺陷小鼠中常具有胞质分裂素2(DOCK2)基因突变,同时由于DOCK2分子具有调节淋巴细胞运输和Toll样受体信号传导作用,这提高了由于IRF5缺陷引起的小鼠狼疮模型中的某些保护作用反而是由于DOCK2缺陷引起的可能性。因此,很多研究分析了缺乏DOCK2突变的混杂效应的情况下IRF5对B细胞发育、分化中的作用。Yasuda等[17]研究发现在不具有DOCK2突变的MRL/lpr狼疮小鼠中抗核抗体和抗dsDNA自身抗体滴度较低,补体固定,IgG同种型IgG2a,IgG2b和IgG3水平较低,肾脏累积较轻,生存率大大提高,说明IRF5可独立于DOCK2突变发挥重要作用。无论有没有DOCK2突变,在含有RNA的免疫复合物激活TLR7和/或通过合成配体激活TLR7和TLR9后,小鼠B细胞或DC产生IFN-α,IFN-β和IL-6的机制中,IRF5都是必不可少的,而且IRF5是从成熟B细胞向浆细胞转变所需的。
4.4 IRF5在T淋巴细胞中的作用
SLE的特点是B细胞和T细胞对自身抗原失去耐受,导致自身抗体的产生和活性细胞因子的干扰。异常的T细胞帮助自身反应的B细胞,使靶器官损害。一个描述SLE细胞因子谱的研究中发现,活动性SLE患者中的IL-6,IL-12,IL-17,IFN-γ和IL-10水平显著高于健康对照组,而IL-4在系统性红斑狼疮较低,Th1/Th2和Th17+Th1/Th2细胞比率上升[18]。在非自身免疫性环境中,IRF5被证明在Th1免疫反应的发展中起着关键作用,在小鼠中敲除IRF5基因可以减少T细胞转移性结肠炎模型中的肠道炎症,减少Th1和Th17,增加Th2细胞因子[19]。在TH1驱动的体内疾病模型中观察到,IRF5缺陷小鼠症状相对IRF5正常小鼠明显减轻。另外,人巨噬细胞中IRF5的过度表达促进了IL-6,IL-12和IL-23p19的转录,导致共培养的人CD4+T细胞中IFN-γ和IL-17的表达增加,促进T淋巴细胞的增殖和活化,并驱使它们朝向TH1或TH17表型分化[20]。综上,IRF5可调节关键促炎因子影响Th1、Th17细胞的增殖与分化。因此,IRF5可通过影响T淋巴细胞增殖、分化在SLE的发生、发展中起作用。
5 总 结
综上所述,IRF5风险单倍体与SLE易感性相关已经得到了证实,此外,IRF5还与其他的一系列人类自身免疫性疾病的风险增加有关。IRF5对体内多种免疫细胞的活性与功能都有重要的调节功能,还需进一步研究IRF5及其蛋白质的功能与SLE患者的疾病状态之间的关系。总之,了解IRF5相关作用机制,将有利于IRF5作为靶点治疗SLE或其他有关的自身免疫性疾病。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
世界科学技术-中医药现代化(2022年3期)2022-08-22
昆明医科大学学报(2022年2期)2022-03-29
昆明医科大学学报(2021年12期)2021-12-30
现代临床医学(2021年4期)2021-07-31
昆明医科大学学报(2021年6期)2021-07-31
昆明医科大学学报(2021年3期)2021-07-22
中国生殖健康(2020年4期)2021-01-18
肿瘤预防与治疗(2019年6期)2019-07-30
医学研究杂志(2015年12期)2015-06-10
