
L-半胱氨酸功能化的碳量子点为探针快速检测花旗松素
2020-05-21 00:30:00程家维张宇辉杨季冬
光散射学报 2020年4期
程家维,张宇辉,杨季冬
(重庆三峡学院, 重庆 404100)
1 引言
花旗松素(taxifolin)也称二氢槲皮素(dihydroquercetin),化学名为5,7,3′,4′-四羟基二氢黄酮醇,是一种生物类黄酮准维生素P,其分子式为C15H12O7,常温下呈淡黄色粉末,主要来源于落叶树的果肉和许多果实中,如大黄花、葡萄和桔子等[1-3]。花旗松素拥有五个酚羟基的特殊分子结构(如图1),成为目前发现的最良好的天然强效抗氧化剂。有研究表明,其可有效去除人体内的自由基与毒素,具有消炎、抗菌、抗癌、调节免疫力、消除黑色素、改善微循环等生物或药理活性,且无毒、无致畸、无突变,工业上用作食品添加剂[4],是许多珍贵药品及保健品的关键成分。日本学者Fukui 首先从针叶树(Chamaecyparis obtusa) 叶中分离出了花旗松素,其生物活性随后被发现并得到了证实[5]。

图1 花旗松素分子结构图Fig.1 The molecular structure of taxifolin
由于花旗松素难溶于水,致其生物利用率低,在医药和临床上的使用受到很大的限制[6]。同时,花旗松素的提取方法各异,伴生物很多,加之市场需求量大,不少以劣充次,因此发展一种快速检测的分析方法尤为重要。目前,常用的检测方法有,高效液相色谱法(HPLC)[7], 紫外-可见分光光度法(UV-vis)[8],方波伏安法(SWV)[3],毛细管区电泳(CZE)[9],液相色谱-质谱法(UPLC-MS)[2]等,这些方法大都需要样品前处理,成本高、操作复杂耗时[10]。与这些报道相比,共振瑞利散射(RRS)光谱法作为一种高灵敏检测的新技术,以其快捷、简单和高灵敏度等优点越来越受分析工作者青睐[11,12]。
纳米科技是21世纪快速发展的主流科技之一,有"21世纪最有前途的材料"的美称,而旗下的一种新兴碳基材料--碳量子点(CDs)作为一种分析新技术材料更是发展迅猛。合成CDs的粒径一般小于10 nm,具有类似于有机染料和半导体量子点的强荧光发射[13],与传统有机染料和普通量子点相比,碳量子点具有光稳定性好、生物相容性好、低毒、分散等优点[14]。据报道,碳量子点替代其它生化类传感器,或作为荧光指示剂的文章层出不穷[15,16],而结合碳点新技术利用共振瑞利散射(RRS)光谱法作为检测手段却鲜见报道。
1 实验部分
1.1 实验仪器
日立F-4500荧光光谱仪(日本,东京),用于测定实验体系的荧光光谱及RRS光谱,在扫描荧光光谱和RRS光谱时狭缝分别设置为10/10 nm和5/5 nm,同时记录好相应的光谱强度。岛津UV-2700紫外-可见分光光度计(日本,东京),用于体系紫外吸收光谱的记录。FA1104N电子天平称(中国,上海),用于实验中药品试剂的称量。利用Nicolet IS10(美国尼高力)光谱仪测量傅里叶红外光谱,通过Zetasizer Nano S(英国马尔文公司)测试合成CDs的zeta电位值,而CDs的XRD则是在BRUCKER D8(德国布鲁克)的检测下完成的。三信Phs-3C-02型酸度计(中国,上海),主要应用于实验中缓冲溶液pH值的调节。
1.2 试剂药品
主要试剂包括:花旗松素(Taxifolin,98%,阿拉丁试剂有限公司,中国上海)、L-半胱氨酸(L-Cys)、柠檬酸、九水合硝酸铁购于中国上海阿拉丁试剂有限公司;其他的一些无机盐(Cu(NO3)2,Mg(OH)2,NaCl,eta)购于中国上海Sinopharm化学试剂有限公司。花旗松素的配制是先用95%的乙醇溶解,再用二次蒸馏水稀释,体积比控制为1∶9。本实验所用到的所有化学品及化学试剂均为分析纯,实验用水均为二次蒸馏水(电阻率为18.2 Ω·cm-1)。
1.3 实验方法
1.3.1L-半胱氨酸修饰的碳量子点的合成
碳量子点的合成是在Yalin Zhang[17]的制备基础上加以改进,通过简易的水热处理法得到,同时得到了L/D-半胱氨酸功能化的CDs,经过后续实验发现L-半胱氨酸功能化的CDs更稳定,因此本实验采用L-半胱氨酸作为修饰剂。在一个典型的反应过程中,利用适量的柠檬酸(1.92g,0.01 mol)和L-半胱氨酸(2.42 g,0.02 moL)作为前驱体共混于烧杯中,再加入5 mL去离子水使其充分溶解分散,并进行30分钟的超声处理;然后将混合液转到聚四氟乙烯密封高压反应釜中,烘箱设定180℃加热1小时;待反应完成,将反应釜放置于室温中自然冷却,获得深棕色溶液;最后透析五天就可获得棕色的发射强蓝绿色荧光的L型CDs,最后存放于4℃的冰箱中备用。
1.3.2光谱扫描测定
在室温条件下,于10 mL的比色管中依次加入20 μL的L型CDs,一定量的Fe3+,最后再加入适当浓度的花旗松素溶液。用二次蒸馏水将上述混合溶液稀释至刻度,振荡摇匀,并在室温条件下静置25 min。最后,扫描体系紫外可见吸收光谱、荧光光谱(λex=345 nm)及RRS光谱。以ΔF=F-F0和ΔIRRS=I-I0记录荧光和RRS变化值。
2 结果与讨论
2.1 L-半胱氨酸修饰的碳量子点
2.1.1L-半胱氨酸修饰的碳量子点的吸收和光学特性
实验在室温条件下,以柠檬酸和L-半胱氨酸(Cys)作为前驱体,采用水热处理法一步合成了高荧光产率的CDs。如图2所示,L-Cys修饰的CDs的紫外吸收光谱其最大吸收峰位于347.5 nm处。而观察CDs的荧光激发和发射光谱,二者呈现出明显的对称关系。在345 nm激发波长下,得到最大发射峰位波长为419 nm,斯托克位移大于50 nm,表明合成的CDs可以消除背景干扰,具有较好的光学性能。
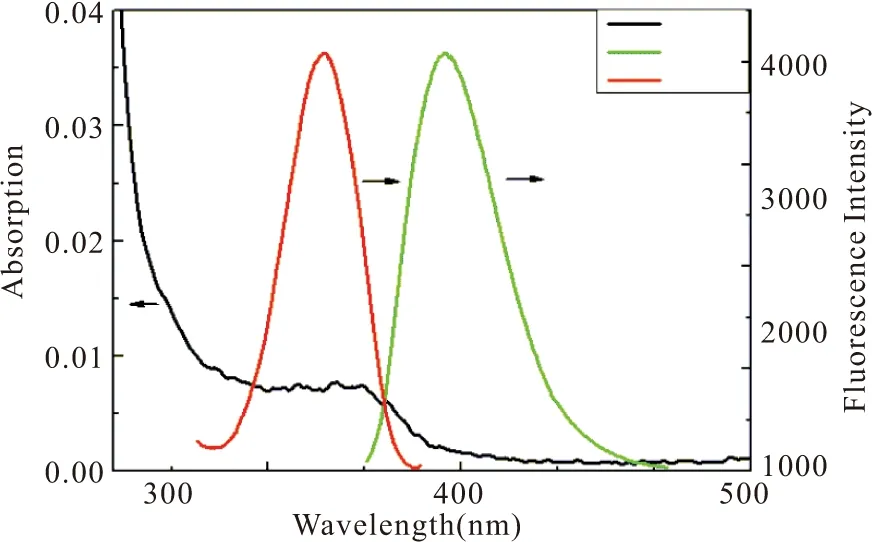
图2 L-半胱氨酸修饰的碳量子点的紫外-可见吸收光谱和荧光光谱图Fig.2 UV-Vis absorption and fluorescence spectra of L-cysteine modified CDs
2.1.2L-半胱氨酸修饰的碳量子点的红外光及其它光学特性
用该法合出的CDs在紫外灯下发射出强的蓝绿色荧光,如图3(a)所示,用X射线衍射(XRD)研究了CDs的晶体结构,可以观察到20°的宽峰,表明其具有明显的非晶结构。为了进一步确认L-Cys修饰的CDs的表面官能团,利用了傅里叶近红外光谱(FT-IR)对其讨论,如图3(b)。3124 cm-1处明显特征峰是由-OH基的拉伸振动引起的,1711 cm-1可归属于C=N的拉伸振动,1585 cm-1处的弱峰很有可能来源于C=C的拉伸振动,而1400 cm-1处的强峰及1225 cm-1处的弱峰都可视为C-N和C-H的变形振动。因此,我们推测半胱氨酸经过水热合成法制备的CDs表面含有大量的-OH、-COOH及-NH等含N/C基团,且具有以水为介质合成CDs的典型特征峰。同时,根据图4发现,制备的水溶性CDs通过Zeta电位测试单峰明显,表明其在水溶液中分散性较好,zeta电位值为-6.60毫伏。
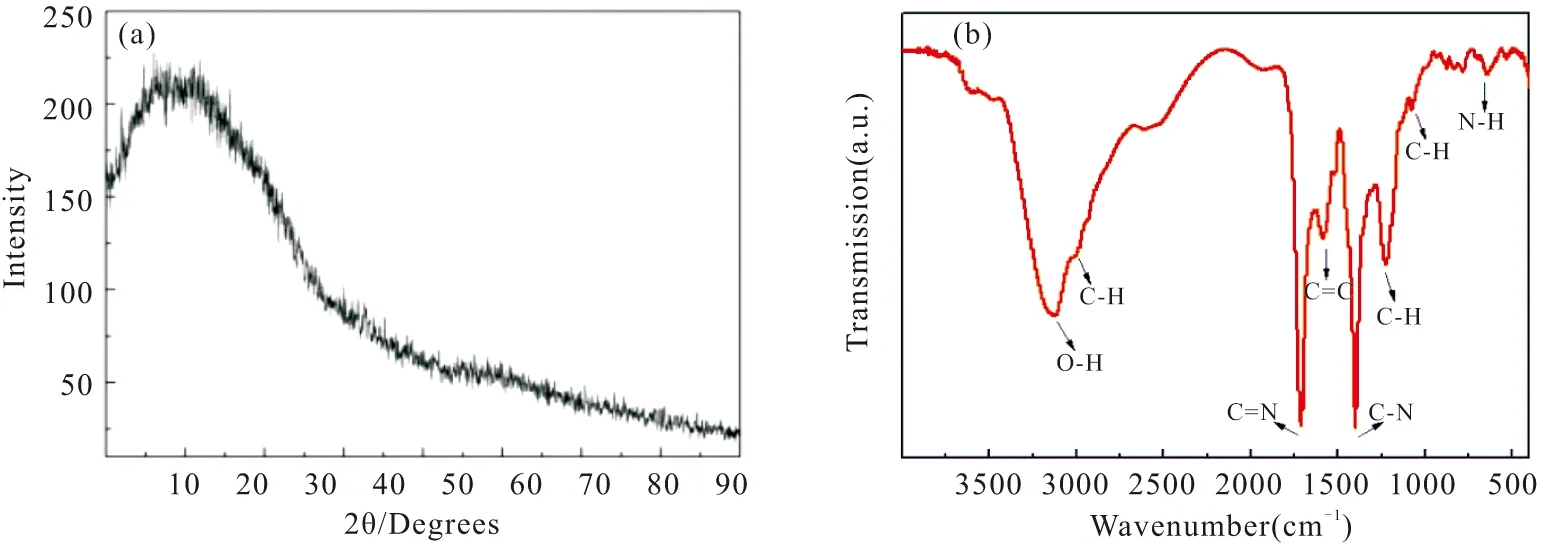
图3 L-半胱氨酸修饰的碳量子点的X射线衍射和傅里叶红外分光光谱Fig.3 XRD and FT-IR spectra of L-cysteine modified CDs

图4 L-半胱氨酸修饰的碳量子点的zeta电位图Fig.4 zeta potential result of L-cysteine modified CDs
2.2 花旗松素对CDs-Fe3+体系的光谱影响
实验研究了CDs、Fe3+及花旗松素三者之间的相互作用,发现三者可以相互反应形成金属螯合物,进而引起体系荧光、吸收和RRS光谱的一系列变化。如图5所示,扫描体系全段的荧光光谱图,发现在350-500 nm波长范围内Fe3+、花旗松素本身是不具荧光特性的,而合成的碳量子点具有强烈的荧光特性,以345 nm为激发波长,在419 nm处出现一个较强的荧光发射特征峰。当把一定量的Fe3+加入到CDs中,体系的荧光信号被显著猝灭,而有趣的是,再向体系中加入一定量的花旗松素,猝灭的荧光竟又得以恢复,形成荧光“开-关”的模式。体系中,花旗松素的浓度与恢复的荧光信号强度在一定浓度范围内存在良好的线性关系,据此建立了分析测定花旗松素的分子荧光光谱法。

图5 三元体系的荧光光谱图Fig.5 Fluorescence spectrum of the CDs-Fe3+-taxifolin.Conditions: CDs, 20 μL; Fe3+, 4×10-5 mol/L; taxifolin:6 μmol/L
如图6所示,在最佳实验条件下扫描体系RRS光谱发现,单独的CDs,Fe3+和花旗松素以及两两存在CDs-Fe3+和CDs-花旗松素时的RRS强度均十分微弱,但Fe3+与花旗松素共存时其RRS强度有一定程度的增加,推测Fe3+与花旗松素的五个酚羟基发生了络合反应形成金属配合物。进一步实验发现CDs,Fe3+和花旗松素三者共存时体系的RRS强度显著增强,这可能是由于三者相互作用形成离子缔合物而引起的,并在381 nm处出现了一个明显的特征峰,RRS强度的变化与花旗松素浓度的增加在一定范围内呈线性关系。后经实验证明,利用Fe3+与花旗松素相作用,线性关系不明显,且没有CDs-Fe3+检测花旗松素构成的三元体系灵敏度高。据此成功建立一种快捷、灵敏检测花旗松素的RRS光谱方法。
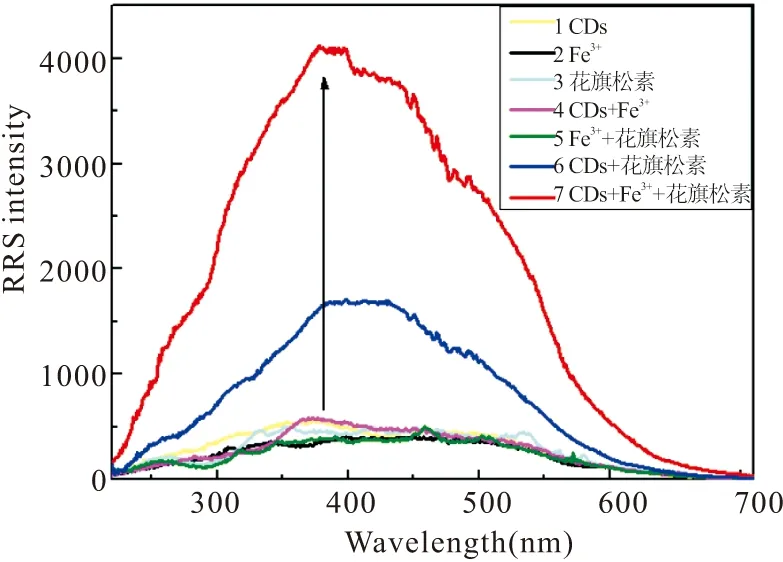
图6 三元体系的共振瑞利散射光谱图Fig.6 RRS spectra of the CDs-Fe3+-taxifolin
Conditions: CDs, 20 μL; Fe3+, 4×10-5mol/L; taxifolin: 6 μmol/L
2.3 实验条件优化
2.3.1缓冲溶液的选择
在实验中,经pH计测定,体系整个环境均处于弱酸态,pH值在3.80-5.72范围内。针对缓冲溶液的选择,主要考察了pH值为3.6时部分酸性缓冲溶液(甘氨酸-盐酸,乙酸-乙酸钠,柠檬酸-柠檬酸钠等),pH值为7.2时常见中性缓冲溶液(HEPES,Tris-HCl,PBS等),以及在不添加缓冲溶液的情况下分别观察体系RRS光谱强度的变化,如图7所示。实验结果发现,不同种类的缓冲溶液对体系的影响不同,酸性环境中的ΔIRRS比中性条件下的大且光谱图有明显的红移现象,但ΔIRRS却在不添加任何缓冲溶液时呈现出最大值且RRS光谱较稳定,从而推测整个实验体系是较稳定的,这也是采用RRS技术作为检测法鲜有的案例。最终,整个实验体系按照其原始的弱酸态(pH=3.80-5.72)进行后续研究,体系本身具有一定的稳定性,不再需要加入缓冲溶液。
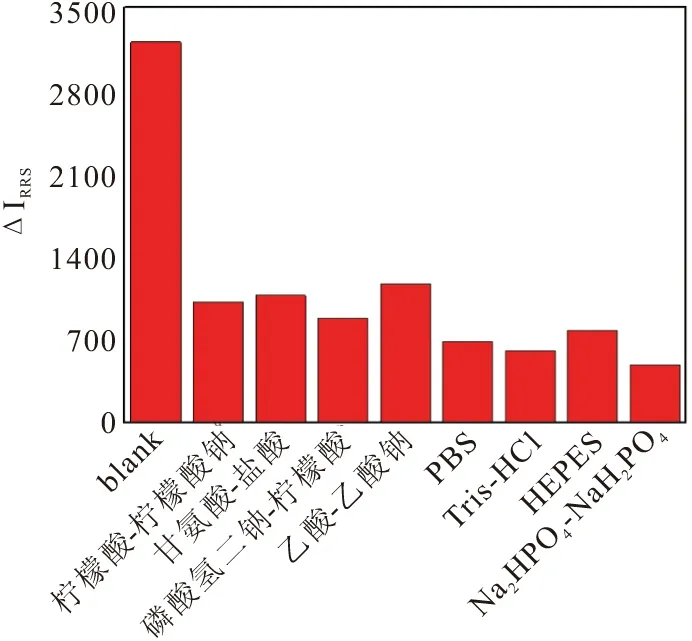
图7 缓冲溶液的选择Fig.7 The choice of buffers kinds. CDs, 20 μL; Fe3+, 4×10-5 mol/L
taxifolin: 6 μmol/L in different buffers, respectively
2.3.2Fe3+浓度的优化
对于整个实验体系而言,Fe3+扮演着非常重要的作用。为了提高体系的灵敏度和一个较宽的线性范围,对体系中Fe3+的浓度进行了优化。根据实验现象,在控制其他条件不变的情况下,随着体系中Fe3+量的逐渐增加,ΔF及ΔIRRS变化显著。经过实验优化,利用分子荧光光谱法测定体系时Fe3+的量确定为5×10-5mol/L。RRS光谱技术中Fe3+量的优化如图8所示,当Fe3+的浓度达到4×10-5mol/L后,体系的ΔIRRS取得最大值。但此时若继续增加过量的Fe3+,可以发现其RRS光谱是不稳定的且ΔIRRS也有明显减小的趋势,这可能是由于过量的Fe3+以游离态的形式先与花旗松素的酚羟基发生络合反应生成金属配合物,从而减小了与CDs表面功能团的竞争性结合,导致该方法的灵敏度不够。反之若进一步减小Fe3+的量,很明显反应不完全,且ΔIRRS会相应的减少而导致整个体系的线性范围变窄,不利于实际样品的分析应用。因此,综合考虑且结合RRS光谱图,最终确定Fe3+的浓度为4×10-5mol/L。
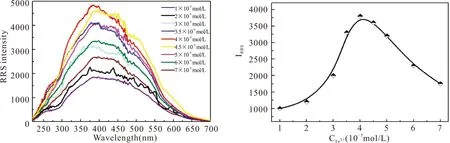
图8 Fe3+浓度的优化Fig.8 (a) RRS spectra of CDs in the presence of Fe3+, (b) ΔIRRS of CDs in different amounts of Fe3+
2.3.4乙醇的影响
由于天然植物活性成分花旗松素是较难溶于冷水中的,因此本实验采用了95%的乙醇对其先进行溶解,待充分溶解后再用二次蒸馏水将其稀释及定容。所以,在后续实验中将进一步考虑乙醇溶液是否会对该体系的现象造成影响。将95%的乙醇溶液按照不同体积的量分别加入到CDs-Fe3+检测花旗松素的实验体系中,记录体系RRS强度变化。如图9所示,在最佳实验条件下,随着乙醇溶液量的增加,体系的ΔIRRS变化不大。故可推测,乙醇溶液对该体系的荧光信号基本无影响。

图9 乙醇的影响Fig.9 Effect of ethanol volume
Conditions: CDs, 20μL; Fe3+, 4×10-5mol/L; taxifolin: 6 μmol/L
2.3.4反应时间及稳定性
为了保证体系中的反应都能够充分进行,在最佳实验条件下,考察了反应时间对体系光谱信号的影响。每数分钟测一次混合体系的RRS光谱并记录ΔIRRS,从而确定反应体系的最佳反应时间及其稳定性。实验记录从5 min开始观察现象,如图10所示,反应一开始是较剧烈的,直到20 min后反应趋于平缓,25 min后基本达到饱和态,且体系的ΔIRRS在1.0 h内趋于稳定。故体系的最佳反应时间选择为25 min,实验测定要求在1.5 h内完成。

图10 反应时间的优化Fig.10 The reaction time and stability
Conditions: CDs: 20μL; Fe3+: 4×10-5mol/L; taxifolin:6 μmol/L
2.4 CDs-Fe3+-花旗松素体系的光谱响应特性
在最佳实验条件下,重复1.3部分的实验方法,记录体系的分子荧光光谱及RRS光谱。由图11(a)可知,将不具荧光特性的Fe3+加入到CDs中,体系的荧光信号被显著猝灭,但当加入同样不具荧光特性的花旗松素后,发生了有趣的一幕,体系中猝灭的荧光信号竟得以恢复。经计算,恢复的荧光强度与花旗松素的浓度在5-150×10-7mol/L范围内呈线性关系。但是,鉴于该方法的灵敏度较低,同时讨论了整个实验体系的ΔIRRS与花旗松素浓度的关系。
如图11(b)所示,波长范围220-800 nm内随着花旗松素浓度的增加,RRS光谱在381 nm特征峰处的散射强度也逐渐增强,且ΔIRRS与花旗松素的浓度在0.9-160×10-7mol/L范围内具有良好的线性关系,最低检出限为8.6×10-9mol/L,线性方程为ΔIRRS=45.67C花旗松素+454.87,相关系数为0.9975。对比荧光光谱法和共振瑞利散射光谱法的线性范围及检出限情况,综合考虑,最终选择更加灵敏且线性范围较宽的RRS光谱法用于实际样品中花旗松素的痕量测定。同时,将RRS光谱法测定花旗松素的相关参数与众多文献相比较,该方法简单、灵敏且快捷,在实际应用中具有较大推广潜值。

图11 荧光光谱响应特征和共振瑞利散射光谱响应特征Fig.11 Fluorescence spectral response characteristics and RRS spectral response characteristicsConditions: CDs, 20μL; Fe3+, 4×10-5 mol/L; taxifolin, 6 μmol/L.
2.5 体系反应机理的推测
2.5.1三元体系之间的反应
如图5所示,Fe3+和花旗松素对CDs荧光信号的影响。CDs在345 nm处有较强的荧光特征峰,在最佳实验条件下,当加入5×10-5mol/L的Fe3+,体系的荧光强度显著猝灭。由于合成碳点的表面含有大量的羟基、羧基及含C、N基团,故可以推测,体系荧光猝灭是由于Fe3+与CDs表面基团发生相互作用形成配位化合物。另外,Fe3+作为过渡金属离子,其顺磁性也可能通过分子内能量转移来猝灭荧光。如图12所示,当Fe3+加入到CDs中,二元体系的吸收光谱带强度增强,从而进一步佐证两者之间发生了反应。

图12 体系的吸收光谱图Fig.12 The UV-Vis absorption spectra of CDs-Fe3+-taxifolin
Conditions: CDs, 20 μL; Fe3+, 4×10-5mol/L; taxifolin, 6 μmol/L.
当花旗松素加入到CDs-Fe3+体系中,体系的荧光得以恢复、RRS信号放大且紫外-可见吸收光谱带强度明显增加。根据荧光光谱、RRS光谱以及紫外-可见吸收光谱的变化,综合考虑这可能是由于花旗松素拥有五个酚羟基的特殊结构处于弱酸态易与金属配合物相结合,加之Fe3+与酚羟基的显色反应,因此可以推断CDs、Fe3+和花旗松素三者以Fe3+为媒介两两作用,最终形成了一种新的金属螯合物。整个实验体系的反应现象简图如图13所示。
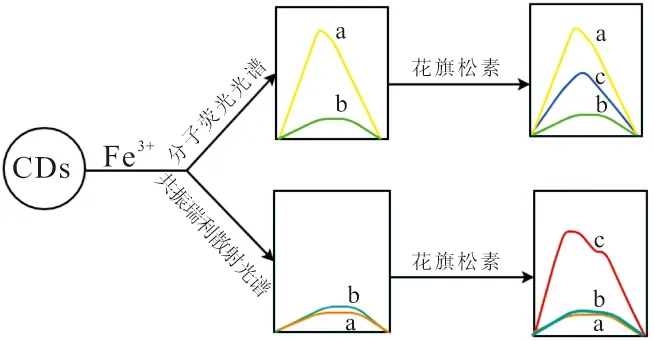
图13 三元体系实验现象图Fig.13 ternary system experimental phenomenon diagram
如图14所示,L-Cys功能化的CDs在与Fe3+作用后,原本在3124cm-1处的O-H特征峰发生了偏移,在3430 cm-1周围形成了-OH宽峰,同时也是以水为介质形成碳点的典型特征峰;而CDs-Fe3+在2424 cm-1出现了一个新的特征峰,而1711 cm-1处的C=N拉伸振动及1225 cm-1处的C-H变形振动消失,原本处于1585 cm-1处的-NH2发生偏移出现在1632 cm-1。对于体系CDs-Fe3+-花旗松素,3430 cm-1周围的O-H拉伸振动变宽,在1263 cm-1处产生了C-H变形振动的新峰,1167 cm-1峰源于C-O拉伸振动,1087 cm-1峰可识别为C-N的变形振动。综上,均可以进一步佐证前面的推测,即三元体系之间相互反应最终生成新的金属螯合物。
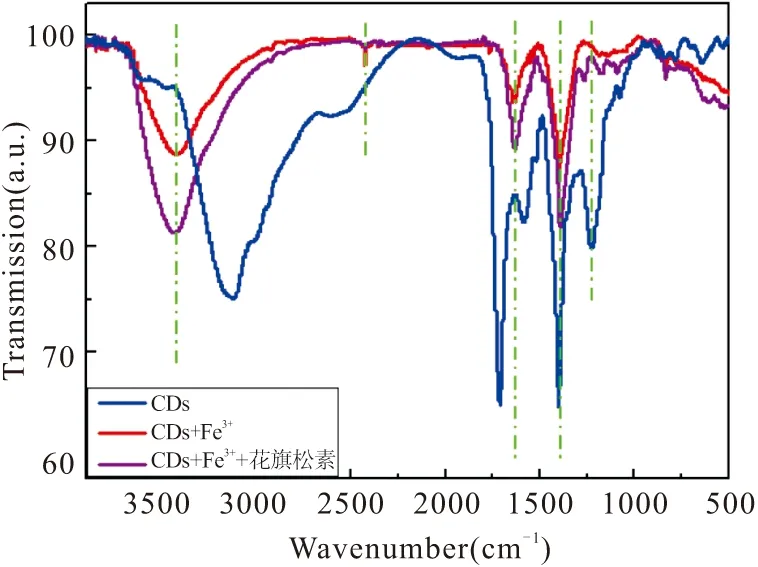
图14 傅里叶红外光谱图Fig.14 The fourier transform infrared(FT-IR) spectrum of ternary system
2.5.2体系荧光和RRS强度变化的原因探索
据文献报道,当共振瑞利散射峰位于或接近于其分子吸收带时,散射通过共振吸收光的能量而发生再散射过程会导致RRS强度增加[18,19]。通过比较吸收光谱和RRS光谱(如图15)发现,RRS光谱特征峰与分子的特征吸收带十分接近,产生再散射过程,导致体系的RRS信号放大,强度显著增强。
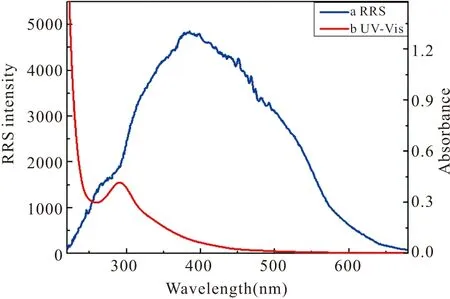
图15 RRS光谱与吸收光谱比较Fig.15 RRS spectrum and absorption spectrum comparison
研究表明,体系中分子体积的大小改变是影响RRS强度的一个重要因素[20,21]。在最佳实验条件下,合成碳点在水溶液中呈单分散状,其本身RRS强度较弱。体系中不存在花旗松素时,CDs-Fe3+的分子质量较小,体积还不够大故RRS强度变化不明显。但是当三者共存时,两两之间通过Fe3+为媒介,形成了新的金属螯合物,分子体积增大,使三元体系CDs-Fe3+-花旗松素的RRS强度显著增强。
另一方面,影响RRS强度变化的因素还要考虑分子的刚性结构[22,23]。在本实验中,Fe3+能够与CDs通过结合位点形成金属配合物,而Fe3+与花旗松素中的酚羟基发生络合反应,三者之间以Fe3+为媒介两两反应最终生成一种新的金属螯合物。通过反应,体系的分子体积增大,同时有可能导致分子的刚性结构发生改变。所以,我们推测体系RRS强度增强的另一个重要原因是由于体系中分子的平面刚性结构改变所致。
2.6 样品分析
采用电子天平准确称取0.0304 g花旗松素,先用95%的乙醇充分溶解再转移至100 mL的容量瓶中用二次蒸馏水定容。实验中利用100 μL的移液枪取60 μL样品溶液于10 mL的比色管中,重复1.3实验方法的平行测定5次,计算纯度。并与出售方标注的高效液相色谱法(HPLC)测定值相比较,如表1所示。再以收集的长江水样做回收实验,将取得的长江水过滤处理,除去一些悬浮的较大杂质,然后3000 rpm转速离心处理30分钟后,取上清液于分析测定。测定方法按照1.3部分,每种浓度平行测定5次,通过标准加入法进行回收实验,将实验结果列于表2中,结果令人满意。

表1 花旗松素的测定结果Table.1 Results for determination of Taxifolin(n=5)
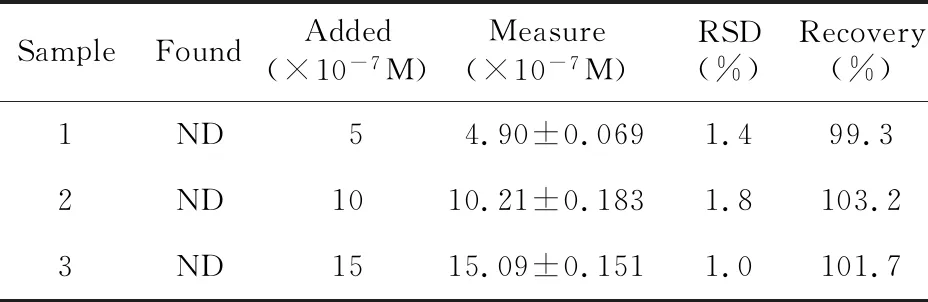
表2 回收率的测定结果Tabl 2 Determination of recovery rate in real samples(n=5)
3 结论
花旗松素作为一种市场稀缺的天然植物活性成分,应用广泛。建立一种快捷、灵敏的痕量检测方法至关重要。文章在最佳实验条件下,记录了体系的RRS光谱、荧光光谱以及紫外-可见吸收光谱来讨论L-Cys修饰的CDs、Fe3+和花旗松素三者之间发生的复杂反应。本文通过考察反应条件、影响因素和机理推测,在最佳实验条件下,成功建立起一种快捷、灵敏痕量检测花旗松素的共振瑞利散射光谱法,ΔIRRS与花旗松素的浓度在0.9-160×10-7mol/L范围内线性变化,检出限为8.6×10-9mol/L。目前采用RRS光谱技术来分析检测天然植物活性成分花旗松素的文献鲜有报道,并且整个实验体系在没有添加缓冲溶液的情况下具有较好的稳定性,也是RRS光谱法作为检测技术出现的少有案例,因此本项工作对RRS作为分析检测的新技术应用具有一定的指导意义。
猜你喜欢
化学与粘合(2020年6期)2020-03-08 09:06:30
现代营销(创富信息版)(2018年7期)2018-09-05 03:24:36
现代营销(创富信息版)(2018年2期)2018-02-10 05:20:49
澳门月刊(2017年12期)2017-12-15 20:45:30
科技视界(2017年25期)2017-12-11 20:30:32
现代营销(创富信息版)(2016年11期)2016-08-22 02:36:20
现代检验医学杂志(2016年5期)2016-08-20 03:17:14
中国当代医药(2015年22期)2015-03-01 02:05:16
安徽医专学报(2014年6期)2014-03-20 13:08:05
河南科技(2014年15期)2014-02-27 14:12:29
