
超声-酶提取毛麝香工艺优化及抗氧化活性研究
2020-05-20 03:29王呈文蔡映举范佳伟
安徽农业科学 2020年8期
王呈文 蔡映举 范佳伟

摘要 [目的]优化毛麝香全草的超声-酶辅助法提取工艺,并评价其抗氧化活性。[方法]采用正交试验优选最佳工艺。运用 DPPH法、ABTS法、NADH - PMS - NBT反应体系法和FRAP法测定总抗氧化能力4种体外抗氧化活性检测方法,并以VC为阳性对照,对比研究毛麝香4种不同提取物的抗氧化活性。[结果]最优工艺条件为乙醇浓度75%、料液比1∶40(g∶mL)、超声温度60 ℃、超声时间30 min、纤维素酶用量0.6%,此条件下的提取率为3.48%。毛麝香乙酸乙酯相的抗氧化能力最佳,对清除3种自由基(DPPH·、ABTS+·、O2·-)的IC50分别为126、156和512 μL,总抗氧化能力测定值为16.7 mg/g。[结论]该提取方法简便、可行,提取物具有良好的抗氧化能力。
关键词 毛麝香;超声-酶辅助;工艺优化;抗氧化
Abstract [Objective] The research aimed to optimize the ultrasonicenzymeassisted extraction process of Adenosma glutinosum and evaluate its antioxidant activity. [Method]The best process was preferred using orthogonal test.Four methods of in vitro antioxidant activity detection by DPPH method, ABTS method, NADHPMSNBT reaction system method and FRAP method were used, and VC was used as a positive control. The antioxidant activities of four different extracts of Adenosma glutinosum were compared. [Result]The optimal process conditions were obtained as follow: ethanol concentration of 75%, solidliquid ratio of 1∶40 (g∶mL), ultrasonic temperature of 60 ℃, ultrasonic time of 30 min and cellulase dosage of 0.6%. The extraction rate of this condition was 3.48%. The ethyl acetate fraction of Adenosma glutinosum had the strongest antioxidant capacity. The IC50 of ethyl acetate fraction for removing three kinds of free radicals (DPPH·, ABTS+·,O2·-) was 126, 156 and 512 μL, respectively, and the total antioxidant capacity was 16.7 mg/g.[Conclusion]The extraction method is simple and feasible, and the extract exhibited strong antioxidant activity.
Key words Adenosma glutinosum; Ultrasonicenzymeassisted; Process optimization; Antioxidant activity
毛麝香[Adenosma glutinosum(L.)Druce]為玄参科毛麝香属植物,属直立本草,别名麝香草、凉草、五凉草、酒子草、毛老虎、饼草、香草(广西)等,分布在我国南部,是一种药用性较强的适应性野生药用植物,具有杀菌、消炎、祛风止痛、散瘀消肿、解毒止痒等功效。有学者对毛麝香进行了挥发油化学成分的研究及乙醇提取物化学成分的研究,并分离得到23个化合物。
目前对毛麝香的基础性研究还不够深入和全面,产业化发展程度较低,主要集中为种植及普通的干草药物销售上,而一些高附加值的深加工产品目前尚未开发,没有充分发掘其价值[1-6]。
超声-酶辅助提取法是一种较新的提取技术,快速方便可行。超声提取的主要理论依据是超声的空化效应、热效应和机械作用。当大能量的超声波作用于介质时,介质被撕裂成许多小空穴,这些小空穴瞬时闭合,并产生高达几千个大气压的瞬间压力,即空化现象。超声空化中微小气泡的爆裂会产生极大的压力,使植物细胞壁及整个生物体的破裂在瞬间完成,缩短了破碎时间,同时超声波产生的振动作用加强了胞内物质的释放、扩散和溶解,从而显著提高提取效率。纤维素酶能够破坏细胞壁,增加溶剂穿透力,高效快速提取细胞内物质,提高了传质速率,对提取物的结构和活性不产生影响,从而提高提取率和缩短提取时间。
目前,对于毛麝香的超声-酶辅助法提取工艺及其抗氧化活性的系统研究鲜见相关报道。该试验通过设计5因素5水平正交试验,对毛麝香的超声-酶辅助法提取工艺进行优化,并进行4种不同自由基的系列抗氧化活性的测试,为进一步科学合理地利用和开发毛麝香的药用价值提供理论依据。
1 材料与方法
1.1 试材
毛麝香,采购自广东汕头药材市场,产地为广东潮汕地区;DPPH·、纤维素酶(400 U/mg),福州飞净生物科技有限公司;ABTS+·、NBT、PMS、NADH、VC标准品,上海如吉生物科技发展有限公司;总抗氧化能力检测试剂盒(FRAP法),上海碧云天生物技术有限公司;其余试剂均为国药或西陇的分析纯。
1.2 仪器
TU-1810紫外可见分光光度计,北京普析通用仪器有限责任公司;DNM-9602自动酶标分析仪,北京普朗新技术有限公司;超声波清洗器,昆仑市超声仪器有限公司;真空冷冻干燥器,杭州聚同电子有限公司;万分之一电子分析天平,赛多利斯(Sartorius)科学仪器(北京)有限公司;大龙移液枪及枪头;其余仪器均为实验室常用试验分析仪器。
1.3 方法
1.3.1 超声-酶辅助法提取毛麝香工艺优化。
1.3.1.1 样品溶液的配制。称取1 g粉粹好的毛麝香,采用超声协同纤维素酶辅助提取法对毛麝香抗氧化活性物质进行提取,提取后,用高速离心机分离,取上清液,再使用0.45 μm 微孔滤膜再次过滤,用容量瓶定容至50 mL,待测[7-12]。
1.3.1.2 DPPH·的清除试验。用无水乙醇配制0.10 mmol/L的DPPH·溶液,现配现用并避光[11-15]。
取0.10 mmol/L的DPPH·溶液6 mL,往其中加0.5 mL样品溶液,混合,避光暗处静置30 min后,519 nm处测吸光度A待测样。
取6 mL的无水乙醇,往其中加0.5 mL样品溶液,混合,519 nm处测吸光度A待测样空白对照。
取0.10 mmol/L的DPPH·溶液6 mL,再加0.5 mL的无水乙醇,混合,避光暗处静置30 min后,519 nm处测吸光度ADPPH·空白对照。
按照公式(1)计算各样品溶液的DPPH·清除率,试验重复3次,取平均值。
DPPH·清除率= 1 -(A待测样 - A待测样空白对照) ADPPH·空白对照×100%(1)
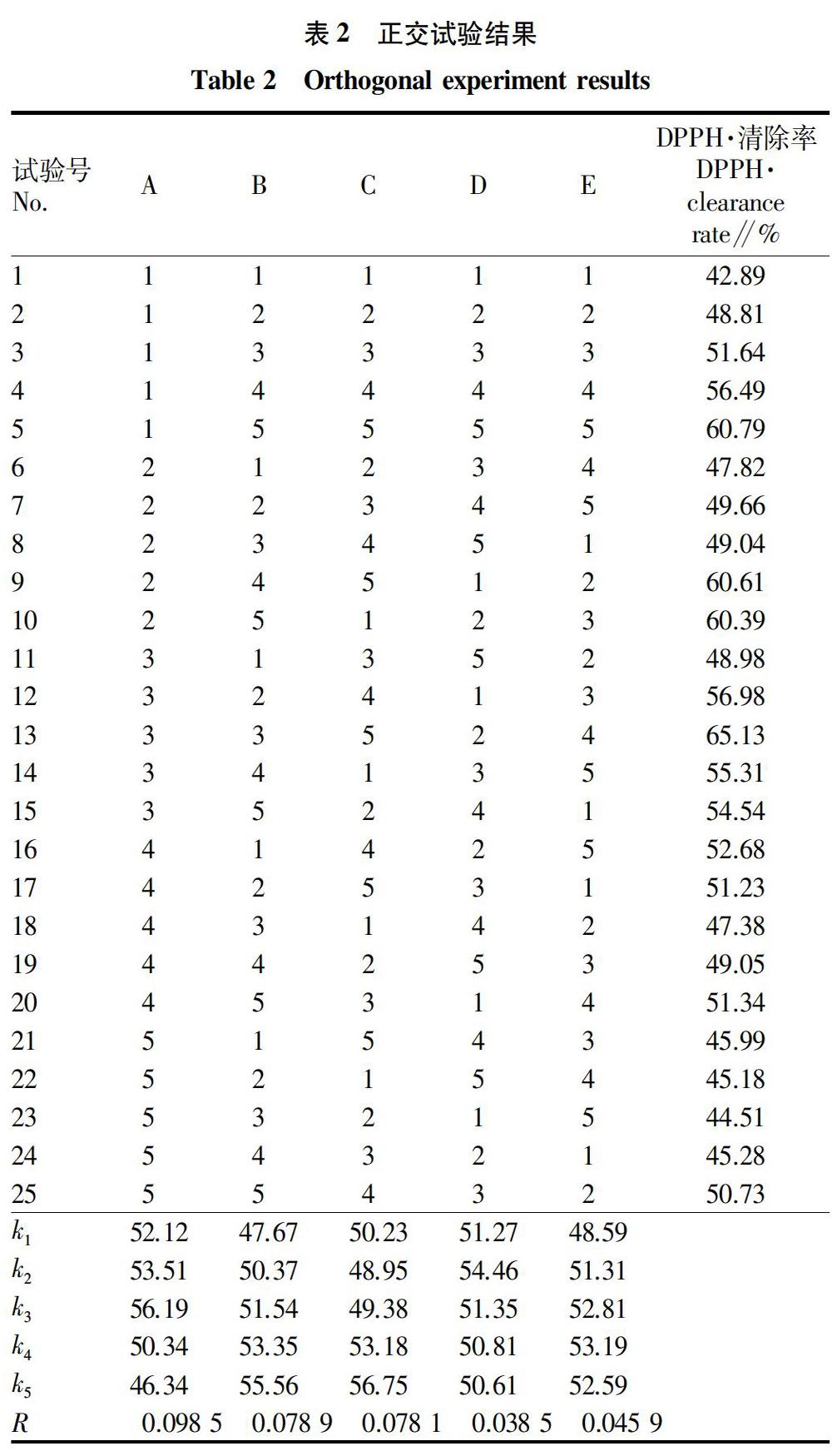
1.3.1.3 正交试验。参考文献并通过预试验,得出乙醇浓度(A)、料液比(B)、超声温度(C)、超声时间(D)、纤维素酶用量(E)是影响毛麝香提取物抗氧化活性的主要因素,故设计5因素5水平用正交表L25(56)安排试验,以DPPH·的清除率为评价指标,筛选最佳提取工艺。
1.3.2 毛麝香提取物的抗氧化活性研究。
1.3.2.1 毛麝香提取物的制备。将毛麝香全草自然晾干,用粉碎机粉碎成粉末,称取毛麝香粉末质量1 kg,按照正交试验的最佳提取工艺条件进行提取,重复3次,合并提取液,旋转蒸发仪减压浓缩得到粗提物浸膏,将部分粗提物浸膏真空冷冻干燥至恒重,待用;另一部分粗提物浸膏用超纯水重新分散溶解,依次用体积比为1∶1的石油醚、乙酸乙酯各萃取5次,分别减压浓缩得到石油醚相浸膏、乙酸乙酯相浸膏和剩余水相浸膏,真空冷冻干燥至恒重,各冻干物冷藏保存待用。
1.3.2.2 样品液的配制。分别准确称取0.010 0 g毛麝香乙醇粗提物、石油醚相、乙酸乙酯相和剩余水相冻干物,用无水乙醇溶解并定容至10 mL,配制成1 mg/mL的样品液,待测。
1.3.2.3 DPPH·清除能力测定。用无水乙醇配制0.10 mmol/L的DPPH·溶液,现配现用并避光[11-14]。
取0.10 mmol/L的DPPH·溶液 4 mL,往其中准确加入x μL不同体积的1 mg/mL 的样品液(根据预试结果设用量梯度),再准确加入(2 000-x)μL的无水乙醇,混合,避光暗处静置30 min后,519 nm处测吸光度A待测样。
准确加入x μL不同体积的1 mg/mL样品液(根据预试结果设梯度),再加(6 000-x)μL的无水乙醇,混合,519 nm处测吸光度A待测样空白对照。
取0.10 mmol/L的DPPH·溶液 4 mL,再加2 mL的无水乙醇,混合,避光暗处静置30 min后,519 nm處测吸光度ADPPH· 空白对照。另以x μL 不同体积的1 mg/mL 的VC溶液(根据预试结果设用量梯度)作为阳性对照。按照公式(1)计算各溶液的DPPH·清除率,试验重复3次,取平均值。
1.3.2.4 ABTS+·清除能力测定。用超纯水配制2.45 mmol/L 过硫酸钾和7 mmol/L ABTS+·混合溶液,避光并静置16 h,配制成ABTS+·储备液。临用前用超纯水稀释至吸光度A734 nm=0.70±0.02 的ABTS+·测试液[11-14]。
取0.10 mmol/L 的ABTS+·测试液 4 mL,往其中准确加入x μL 不同体积的1 mg/mL 的样品液(根据预试结果设用量梯度),再准确加入(2 000-x)μL 的无水乙醇,混合,避光暗处静置6 min后,734 nm处测吸光度A待测样。
准确加入 x μL 不同体积的1 mg/mL 的样品液(根据预试结果设梯度),再加(2 000-x)μL 的无水乙醇、4 mL超纯水,混合,734 nm处测吸光度A待测样空白对照。
取0.10 mmol/L的ABTS+·测试液4 mL,再加2 mL的无水乙醇,混合,避光暗处静置6 min 后,734 nm处测吸光度AABTS·空白对照。
另以x μL 不同体积的1 mg/mL 的VC溶液(根据预试结果设用量梯度)作为阳性对照。按照公式(2)计算各溶液的ABTS+·清除率,试验重复3次,取平均值。
ABTS+·清除率=[1 -(A待测样 -A待测样空白对照)/AABTS·空白对照]×100%(2)
1.3.2.5 O2·-清除能力测定。用超纯水分别配制0.3 mmol/L的NBT磷酸缓冲液(0.1 mol/L,pH=7.4)、0.936 mmol/L的NADH磷酸缓冲液(0.1 mol/L,pH=7.4)和0.120 mmol/L的PMS磷酸缓冲液(0.1 mol/L,pH=7.4)[14-15]。
依次分别加入1.5 mL的以上3种磷酸緩冲液,再往其中准确加入 x μL 不同体积的1 mg/mL 的样品液(根据预试结果设用量梯度),再准确加入(1 500-x)μL 的无水乙醇,混合,避光暗处静置5 min后,560 nm处测吸光度A待测样。
准确加入x μL 不同体积的1 mg/mL 的样品液(根据预试结果设梯度),再加(1 500-x)μL 的无水乙醇,4.5 mL超纯水,混合,560 nm处测吸光度A待测样空白对照。依次分别加入1.5 mL的以上3种磷酸缓冲液,再加1.5 mL的无水乙醇,混合,避光暗处静置5 min后,560 nm处测吸光度AO2·- 空白对照。
另以x μL 不同体积的1 mg/mL 的VC溶液(根据预试结果设用量梯度)作为阳性对照。按照公式(3)计算各溶液的O2·-清除率,试验重复3次,取平均值。
O2·-清除率=1 -(A待测样 - A待测样空白对照)AO2·-空白对照×100%(3)
1.3.2.6 FRAP 法测定总抗氧化能力。用超纯水分别配制10 mmol/L TPTZ溶液(40 mmol/L盐酸溶液配制)、100 mmol/L醋酸缓冲液(pH=3.6)和20 mmol/L氯化铁溶液,按10∶1∶1的比例进行混匀后得RFAP测试液,现配现用。 取3.9 mL RFAP测试液,加入0.1 mL 0.5 mg/mL的待测样品溶液(浓度根据预试结果设置),充分混匀后,在37 ℃的恒温水浴中反应10 min,于593 nm下测定吸光度,以无水乙醇为空白对照。结果表达为每1 g干重相当于Trolox还原力的相当量(mg Trolox equivalent antioxidant capacity/g dry weight,mg TEAC/g DW),试验重复3次,取平均值[14-15]。
2 结果与分析
2.1 超声-酶辅助法提取毛麝香正交试验
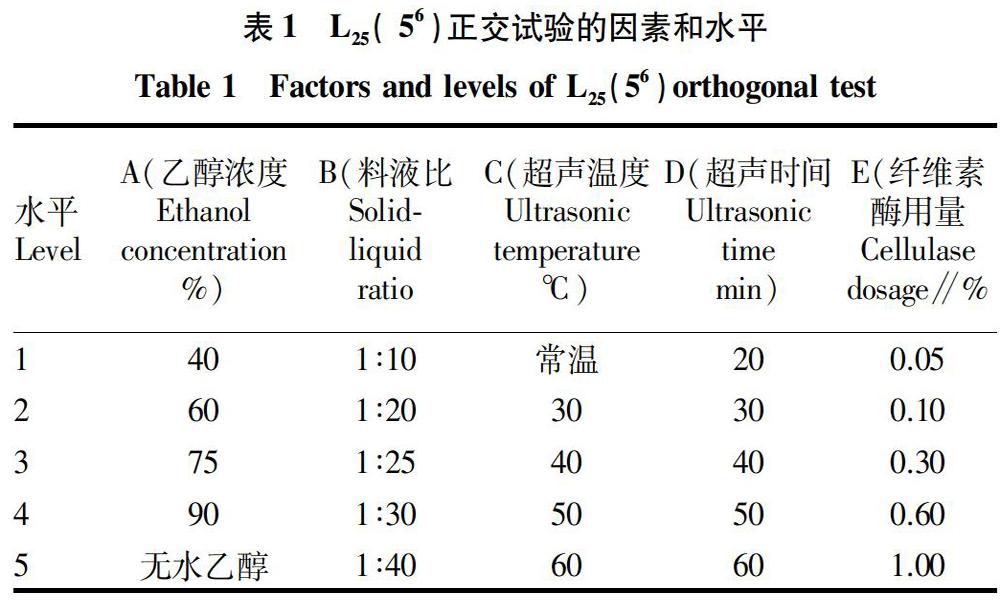
由正交试验的测定结果(表2)可以分析因素影响的次序,R值越大,影响越大,RA>RB>RC>RE>RD,即影响毛麝香提取物抗氧化活性的因素的主次顺序为乙醇浓度(A)>料液比(B)>超声温度(C)>纤维素酶用量(E)>超声时间(D);同时分析k值,可得出最优组合为A3B5C5D2E4,即乙醇浓度75%、料液比1∶40(g∶mL)、超声温度60 ℃、超声时间30 min、纤维素酶用量0.6%,此最佳工艺条件的毛麝香乙醇粗提物的提取率为3.48%。
2.2 毛麝香提取物的抗氧化活性比较
试验结果表明(表3),毛麝香乙醇粗提物及其不同极性组分均有一定的抗氧化能力,其中乙酸乙酯极性组对于清除3种自由基(DPPH·、ABTS+·、O2·-)的IC50 值分别为126、156和512 μL,总抗氧化能力测定值为16.7 mg/g,是4种不同提取物中抗氧化能力最强的组分,但抗氧化能力略低于阳性对照(VC)组。 其余3个提取物在4种抗氧化评价体系中也均表现出相同的变化趋势,抗氧化活性强弱从大到小依次为乙酸乙酯相>乙醇粗提物>剩余水相>石油醚相。乙酸乙酯组分中多为中等极性的成分,其含有较丰富的具有良好抗氧化功效的多酚、黄酮类化合物。
2.3 重复性试验
取5份试样,按照最佳提取工艺进行提取,毛麝香乙醇粗提物的提取率分别为3.38%、3.42%、3.59%、3.48%、3.63%,并用其进行DPPH·清除能力测定,得到的IC50值分别为310、332、308、315、335。
3 结论
超声-酶辅助提取法高效简便,且提高了毛麝香有效成分的提取率,是一种较为理想的毛麝香抗氧化活性成分的提取方法。通过5因素5水平的正交试验,得出影响毛麝香提取物抗氧化活性的因素的主次顺序为乙醇浓度(A)>料液比(B)>超声温度(C)>纤维素酶用量(E)>超声时间(D),以及最优工艺条件为乙醇浓度75%、料液比1∶40(g∶mL)、超声温度60 ℃、超声时间30 min、纤维素酶用量0.6%,此最佳工艺条件下毛麝香乙醇粗提物的提取率为3.48%,而没有加入纤维素酶辅助提取的提取率仅为2.61%。
抗氧化活性试验采用了4种抗氧化评价方法,较为系统客观地对比分析了毛麝香乙醇粗提物及其不同极性组分(乙醇粗提物、石油醚相、乙酸乙酯相、剩余水相)的抗氧化活性,结果表明4种不同提取物均有一定的抗氧化能力,也均表现出相同的变化趋势,抗氧化活性强弱从大到小依次为乙酸乙酯相>乙醇粗提物>剩余水相>石油醚相,其中乙酸乙酯相的抗氧化活性最佳,但抗氧化效果略低于阳性对照(VC)组。
参考文献
[1] 汪存存,卫罡,李润美.毛麝香挥发油成分的GC-MS分析[J].中国中医药信息杂志,2008,15(2):36-37.
[2] 潘文昭.毛麝香的药用功效[J].农村新技术,2013(3):46.
[3] 杨东娟,詹怀先,石磊.毛麝香叶挥发油化学成分研究[J].西北林学院学报,2013,28(2):164-167.
[4] 谭冰心.毛麝香(Adenosma glutinosum(L.)Druce)化学成分研究[D].广州:广州中医药大学,2017.
[5] 谭冰心,彭光天,于思,等.毛麝香的化学成分研究[J].中草药,2017,48(10):2024-2027.
[6] 于思,彭光天,雷玉,等.毛麝香的化学成分研究(Ⅱ)[J].中山大学学报(自然科学版),2018,57(3):89-95.
[7] 包怡红,王硕,王文琼,等.超声波酶法提取红松树皮中多酚类化合物的研究[J].食品工业科技,2013,34(3):232-236.
[8] 王彦平,杨庆莹,孙瑞琳,等.不同酶法辅助提取紫山药皮薯蓣皂苷及其抗氧化活性研究[J].食品工业科技,2017,38(10):200-204.
[9] 张健,刘少伟,张毅,等.仿刺参精酶解工艺条件优化及体外抗氧化[J].食品工业科技,2017,38(5):232-237.
[10] 原姣姣,陈锦璇,张帆,等.响应面优化超声_酶辅助强化油橄榄叶多糖的提取[J].中国油脂,2019,44(4):128-132.
[11] 王呈文,纪明慧,舒火明,等.牛大力总黄酮提取工艺及不同萃取物的抗氧化活性研究[J].化学研究与应用,2013,25(5):713-717.
[12] 王呈文,纪明慧,陈光英,等.热带莫氏兰根提取物的抗氧化活性及稳定性研究[J].食品工业科技,2013,34(5):209-211,217.
[13] 王国良,李建科,吴晓霞,等.水麻果多酚的提取纯化及其抗氧化、抗肿瘤活性作用[J].天然产物研究与开发,2019,31(1):1-9.
[14] GE Y,DUAN Y F,FANG G Z,et al.Polysaccharides from fruit calyx of Physalis alkekengi var.francheti:Isolation,purification,structural features and antioxidant activities [J].Carbohydrate polymers,2009,77:188-193.
[15] 楚秉泉,方若思,李玲,等.洋甘菊各萃取相抗氧化活性及其有效成分分析[J].食品工业科技,2019,40(8):1-6.
猜你喜欢
当代水产(2021年6期)2021-08-13
科学与财富(2016年26期)2016-12-01
中国实用医药(2016年24期)2016-10-17
中国实用医药(2016年24期)2016-10-17
科学与财富(2016年28期)2016-10-14
食品工业科技(2014年15期)2014-03-11
食品工业科技(2014年15期)2014-03-11
食品工业科技(2014年9期)2014-03-11
