
新型钌配合物的制备及在光催化氧化中的应用
2020-05-13 02:58楼江丽
广州化工 2020年8期
楼江丽,曹 媛,张 艳
(江南大学药学院,江苏 无锡 214122)

有机化合物的选择性氧化是指需要在不同环境中区分相同官能团的催化剂或试剂以实现此类结构的选择性直接官能化,是有机合成中最基本的反应之一,也是工业化学中最关键的挑战之一[5]。
β-二酮酸酯是应用配位化学中最常用的配体之一,可与所有金属离子形成稳定的螯合物并具有挥发性,例如应用于金属有机化学气相沉积(MOCVD)以及其他相关技术[6]。尽管它们具有不饱和特性π电子离域,通常不被视为具有氧化还原活性的非纯配体。但是,最近的一份关于NacNac镍络合物的报告证明磁性测量结果是含有氧化的配体的[7],因此我们选择了β-二酮酸酯中具有代表性的9-氧化邻苯二酚(ply)来做钌的配合物。
本文使用钌和ply配合成三种新的光催化剂,将其应用于选择性氧化苄基C-H键到羰基、选择性氧化胺类到醛类以及选择性氧化硫化物到亚砜这三类选择性氧化反应,并在氧化胺类到醛类和氧化硫化物到亚砜这两类选择性氧化反应中获得了中等产率,可将其应用于目前已上市的药物分子的合成中,对其进行生物活性测试,以期找到具有较好生物活性的先导化合物。
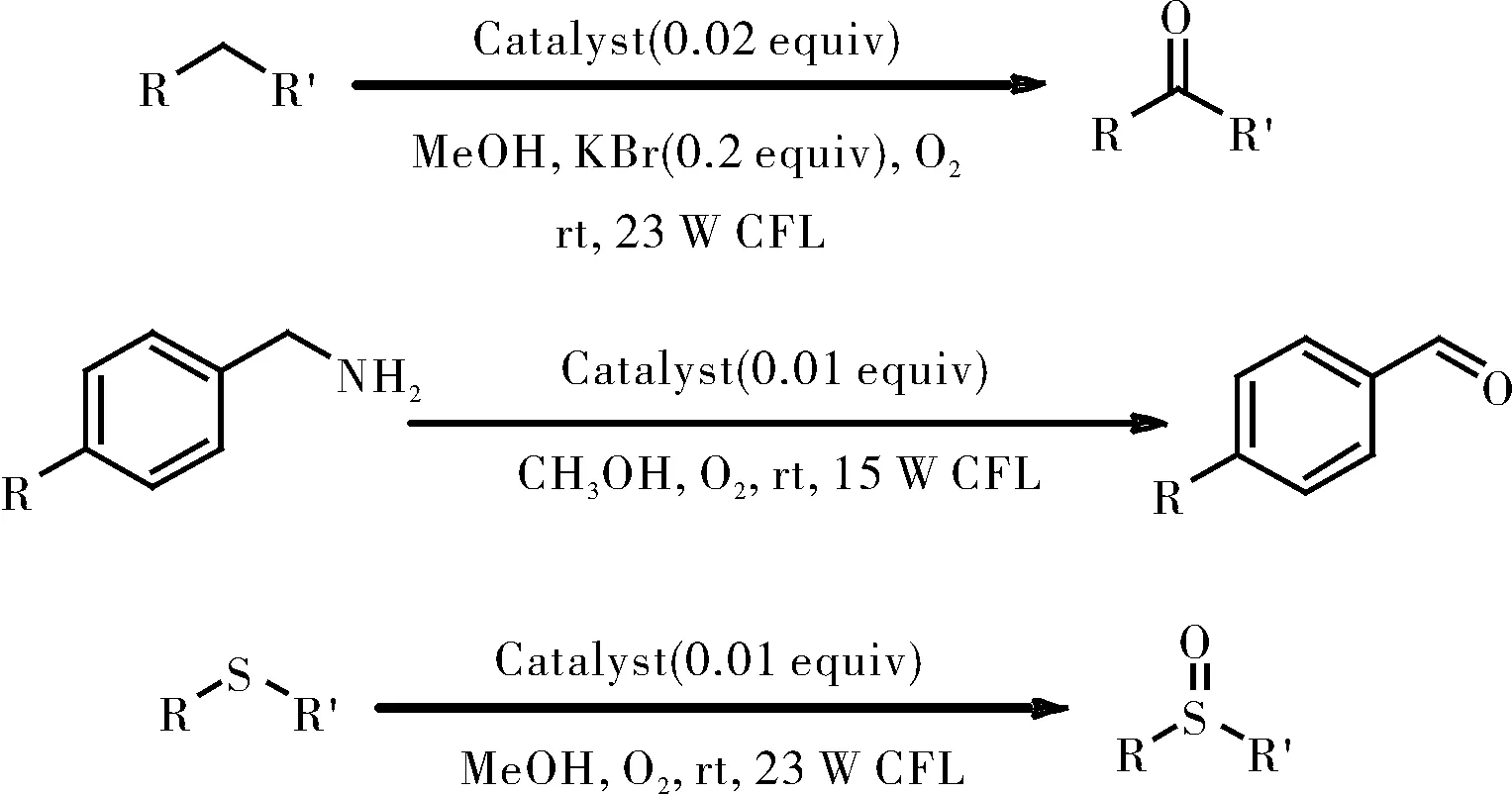
图1 三种配合物催化的选择性氧化反应
1 实 验
1.1 仪器与试剂
旋转蒸发仪、循环水式多用真空泵、集热式恒温加热磁力搅拌器、真空干燥箱,郑州科泰实验设备有限公司;LC-MS,Waters Micromass Platform LCZ Mass Spectrometer(美国);核磁共振仪,Bruker AVⅢ(德国);气相色谱仪,Thermo Fisher Scientific(美国);电子天平,常熟市佳衡天平仪器有限公司;LED灯E27大螺口,飞利浦。
2-甲氧基萘、氯化铝、肉桂酰氯、2,2’-联吡啶、高氯酸银、氯化锂、2-氨基吡啶、苯、亚硝基苯、三水合三氯化钌、三苯基膦乙基苯、硝酸银、对溴乙基苯、对甲氧基乙基苯、茚满、二苯基甲烷、苄胺、异色满、4-甲基苄胺、4-(三氟甲基)苄胺、对氯苄胺、甲基苯基硫醚、二丁基硫醚、苄基苯基硫醚、4-氯茴香硫醚、烯丙氧基苯硫醚、二苯硫醚、苯乙硫醚、二苄基硫醚等原料未经进一步处理,购自萨恩化学技术(上海)有限公司、上海科技医药毕得有限公司和国药试剂有限公司。
1.2 实验方法
1.2.1 9-氧化邻苯二酚的合成
将2-甲氧基萘(0.01 mol)和1当量的肉桂酰氯溶解在1,2-二氯乙烷(100 mL)中。在将反应烧瓶在冰浴中冷却之后,在机械搅拌下将0.5当量氯化铝缓慢加入到烧瓶中。当反应达到室温时,再向溶液中加入20当量氯化铝,并将混合物回流3 h。反应结束后将混合物与冰盐酸混合,过滤。滤液用二氯甲烷萃取。将固体反复用热的二氯甲烷煮沸并过滤直到滤液变为无色。合并所有有机萃取物,用无水硫酸钠干燥,并置于旋转蒸发仪上,得到黄色固体产物9-氧化邻苯二酚(ply)。

图2 9-氧化邻苯二酚的制备
1.2.2 [RuⅡ(ply)(bpy)2]ClO4的制备
1.2.2.1 Ru(bpy)2Cl2·2H2O的制备
将三水合三氯化钌(0.01 mol)和2,2’-联吡啶(0.02 mol)溶解在100 mL二甲基甲酰胺中,在10 min内加入氯化锂(0.02 mol),将所得混合物在氮气氛围下回流8 h。将反应混合物蒸发至干,使用中性氧化铝柱纯化粗品,通过5:1二氯甲烷/乙腈的溶剂混合物洗脱得产物Ru(bpy)2Cl2·2H2O。

图3 Ru(bpy)2Cl2·2H2O的制备
1.2.2.2 [RuII(ply)(bpy)2]ClO4的制备
将0.2 mmol的Ru(bpy)2Cl2·2H2O和0.4 mmol的AgClO4加入30 mL乙醇/水(2:1)中。将所得混合物在氮气氛围下回流1 h。滤出沉淀的AgCl,向滤液中加入0.2 mmol的ply和0.2 mmol 的NEt3。将混合物在氮气氛围下回流6 h后用旋转蒸发仪将反应混合物蒸发至干。通过使用中性氧化铝柱纯化粗产物,通过5:1二氯甲烷/乙腈的溶剂混合物洗脱得产物[RuII(ply)(bpy)2]ClO4。

图4 [RuII(ply)(bpy)2]ClO4的制备
1.2.3 [RuII(pap)2(ply)]ClO4的制备
1.2.3.1 2-(苯基偶氮)吡啶的制备
将2-氨基吡啶(5 g)加入到温热的50%氢氧化钠溶液(50 mL)中,温和加热,然后加入苯(3 mL)。在10 min内加入亚硝基苯(6 g),同时摇动,并将混合物再加热10 min。接着用苯(3×100 mL)萃取,得到溶液,趁热过滤,减压浓缩至100 mL。使用中性氧化铝柱纯化,通过5:1二氯甲烷/乙腈的溶剂混合物洗脱得产物2-(苯基偶氮)吡啶(pap)。
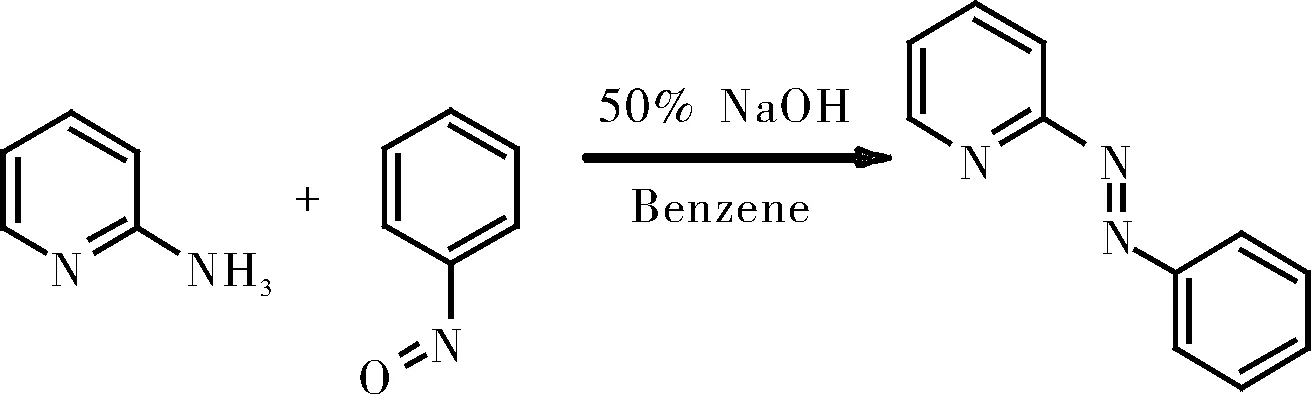
图5 2-(苯基偶氮)吡啶的制备
1.2.3.2 RuCl2(pap)2的制备
在氮气环境下,向溶解在15 mL甲醇中的0.5 mmol的三水合三氯化钌中加入溶解在5 mL甲醇中的pap(1.09 mmol)。将绿色混合物在氮气氛围下加热回流,同时磁力搅拌8 h。将溶液冷却至室温后,过滤收集沉淀物,用水彻底洗涤,最后用乙醚洗涤。使用中性氧化铝柱纯化,通过二氯甲烷/乙腈(5:1)的溶剂混合物洗脱得产物RuCl2(pap)2。
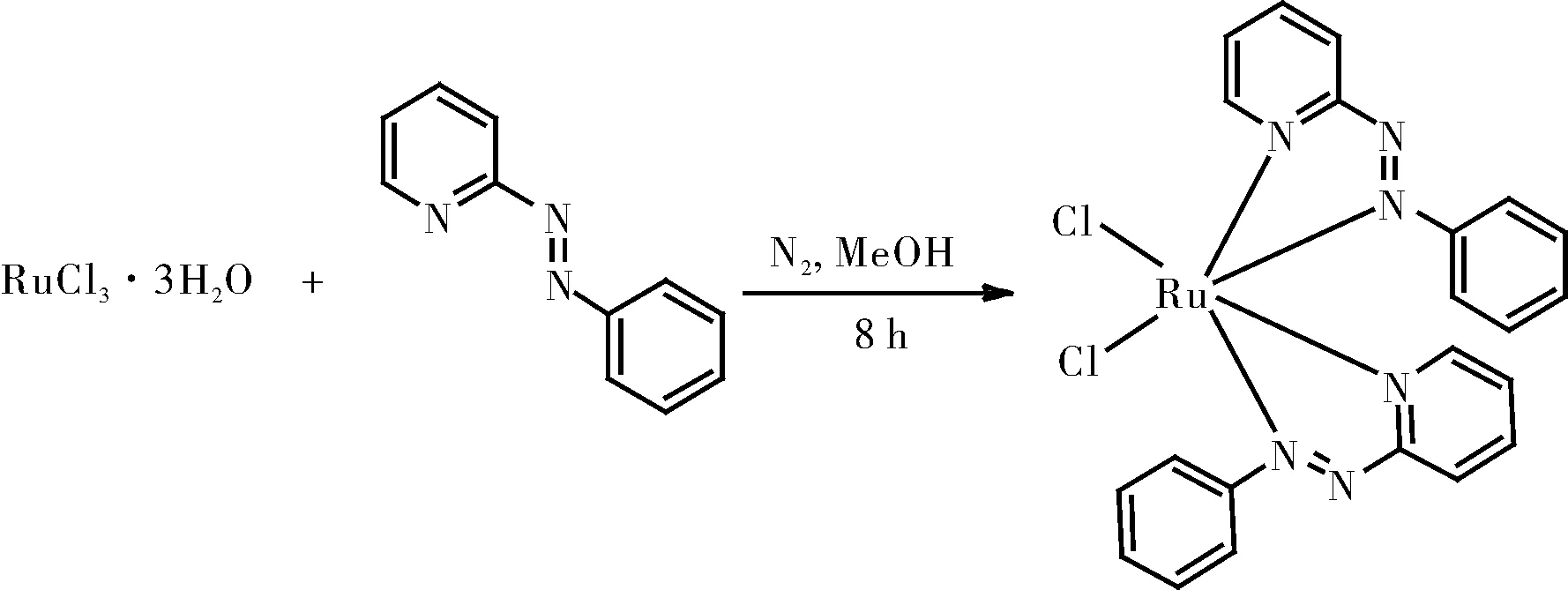
图6 RuCl2(pap)2的制备
1.2.3.3 [RuII(pap)2(ply)]ClO4的制备
将0.19 mmol的RuCl2(pap)2和0.38 mmol的AgNO3在乙醇(30 mL)中的混合物在氮气氛围下加热回流1 h。滤出沉淀的AgCl,并将0.19 mmol的ply和0.2 mmol的NEt3加入滤液中。将混合物在氮气氛围下加热回流6 h。将反应混合物蒸发至干,将粗产物溶于最小体积的MeCN中,并加入饱和NaClO4溶液(10 mL)。滤出深棕色沉淀,用冰冷的蒸馏水洗涤两次,并在真空下干燥。使用中性氧化铝柱纯化粗产物,通过二氯甲烷/乙腈(5:1)的溶剂混合物洗脱得产物[RuII(pap)2(ply)]ClO4。

图7 [RuII(pap)2(ply)]ClO4的制备
1.2.4 RuII(pap)(ply)(PPh3)的制备
1.2.4.1 Ru(PPh3)3Cl2的制备
将三氯化钌三水合物(0.2 g)溶解在甲醇(50 mL)中,加入六倍过量(1.2 g)的三苯基膦,将溶液在氮气氛围下回流3 h。将得到的晶体分别用甲醇和乙醚充分洗涤,真空干燥(60 ℃)得产物Ru(PPh3)3Cl2。
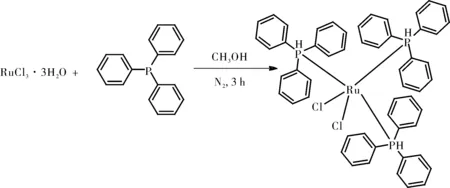
图8 Ru(PPh3)3Cl2的制备
1.2.4.2 Ru(PPh3)2(pap)Cl2的制备
向0.52 mmol的Ru(PPh3)3Cl2和0.55 mmol的pap的混合物中加入二氯甲烷(50 mL)。将所得的红紫色溶液搅拌1.5 h。在该红紫色溶液上层叠己烷(100 mL)。缓慢扩散溶剂,Ru(PPh3)2(pap)Cl2沉积为闪亮的针状晶体,过滤收集,用己烷彻底洗涤,真空干燥得产物Ru(PPh3)2(pap)Cl2。

图9 Ru(PPh3)2(pap)Cl2的制备
1.2.4.3 RuII(pap)(ply)(PPh3)的制备
将0.17 mmol的Ru(PPh3)2(pap)Cl2和0.34 mmol的AgClO4在EtOH(30 mL)中的混合物在氮气氛围下加热回流1.5 h。过滤除去沉淀的AgCl,并将0.21 mmol的ply和0.43 mmol的NEt3加入滤液中。将混合物在氮气氛围下加热回流12 h。将反应混合物蒸发至干,并通过中性氧化铝柱纯化。用二氯甲烷/乙腈(5:1)的混合物洗脱得产物RuII(pap)(ply)(PPh3)。
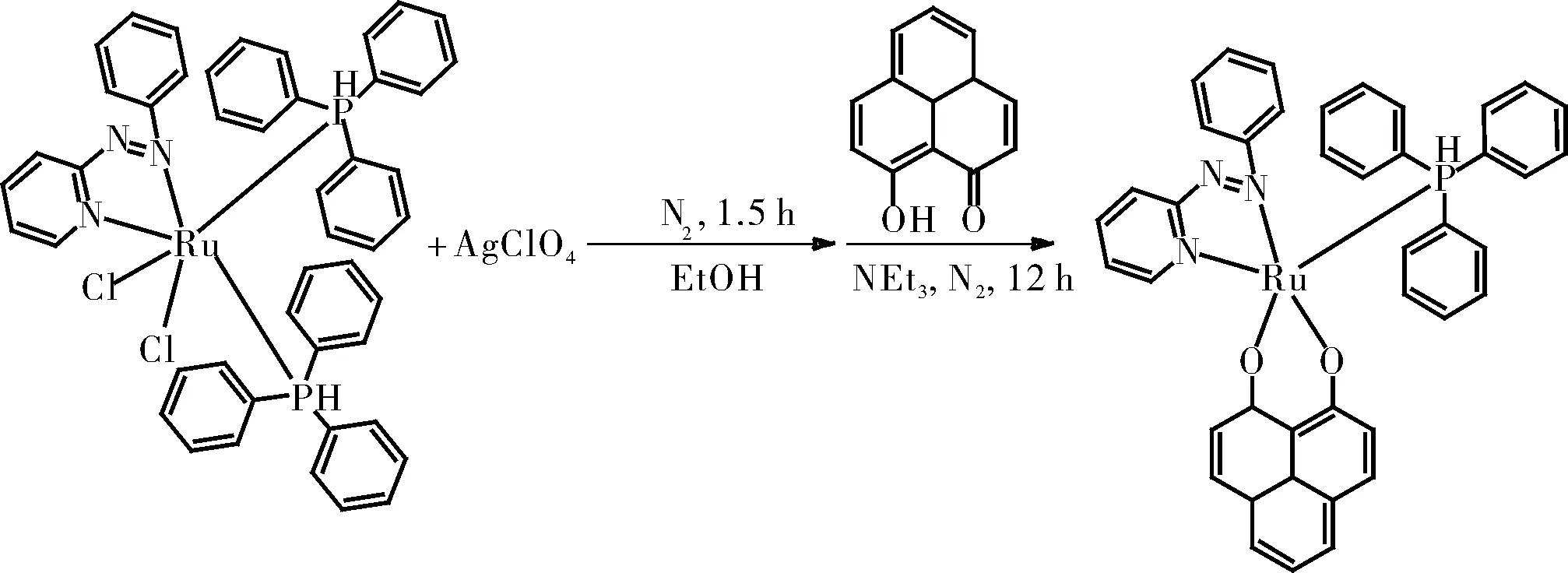
图10 RuII(pap)(ply)(PPh3)的制备
1.2.5 催化苄基衍生物的氧化
在带有磁力搅拌棒的10 mL史莱克管中,将苄基衍生物(0.25 mmol),溴化钾(0.2当量)和催化剂([RuII(ply)(bpy)2]ClO4/[RuII(pap)2(PLY)]ClO4/RuII(pap)(PLY)(PPh3),2mol%)溶于甲醇(2 mL)中。并将所得混合物在O2氛围下置于23 W白色CFL照射8 h,如图11所示。当反应结束后,将反应混合物用盐水洗涤。用乙酸乙酯再次萃取水相,最后合并有机相。有机相经Na2SO4干燥并真空浓缩,所得残余物通过硅胶柱色谱法纯化,得到所需产物。
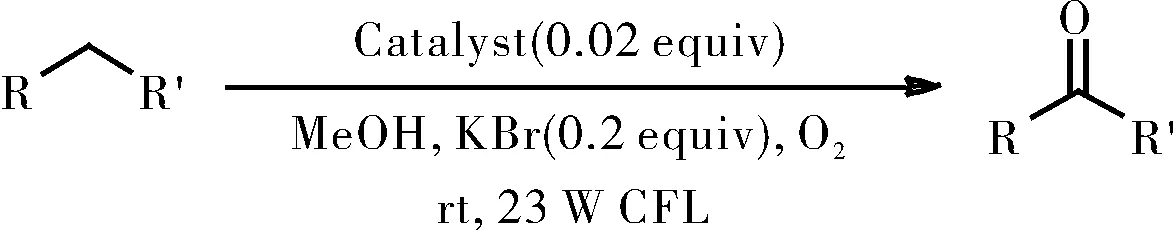
图11 苄基衍生物的氧化
1.2.6 催化胺的氧化
在带有磁力搅拌棒的10 mL史莱克管中,将胺(0.25 mmol)分别和三种改造后的催化剂([RuII(ply)(bpy)2]ClO4/[RuII(pap)2(ply)]ClO4/RuII(pap)(ply)(PPh3),1mol%)溶于甲醇(2 mL)中。并将所得混合物在O2气氛下置于15 W白色CFL照射8 h,如图12所示。通过GC分析监测反应进程。使用二苯基作为内标,通过GC测量计算产物的光氧化率。
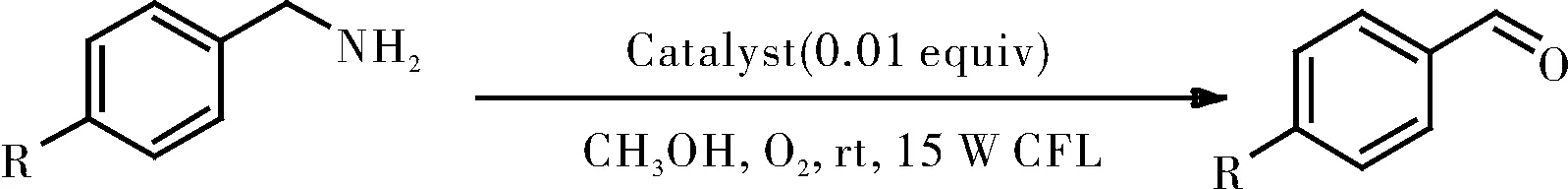
图12胺的氧化
Fig.12 Oxidation of amine
1.2.7 催化硫化物的氧化
在带有磁力搅拌棒的10 mL史莱克管中,将硫化物(0.25 mmol)分别和三种改造后的催化剂([RuII(ply)(bpy)2]ClO4/[RuII(pap)2(ply)]ClO4/RuII(pap)(ply)(PPh3),1mol%)溶于甲醇(2 mL)中。并将所得混合物在O2氛围下置于23 W白色CFL照射8 h,如图13所示。当反应结束后,将反应混合物用盐水洗涤。用乙酸乙酯再次萃取水相,最后合并有机相。有机相经Na2SO4干燥并真空浓缩,所得残余物通过硅胶柱色谱法纯化,得到所需产物。
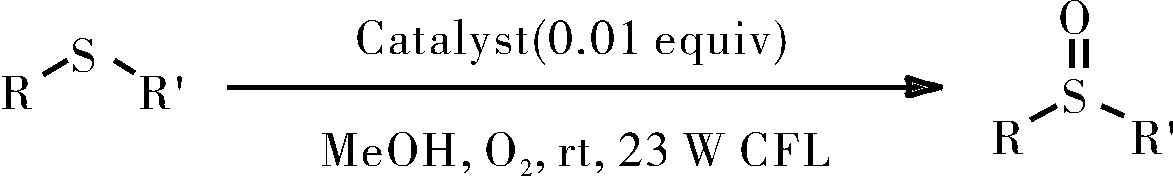
图13硫化物的氧化
Fig.13 Oxidation of sulfide
2 结果与讨论
2.1 三种配合物的合成
按照“1.2实验方法”制备了一系列的9-氧化邻苯二酚和钌的配合物,我们制备了三种配合物,合成每一种配合物的每一步的产率如表1所示。
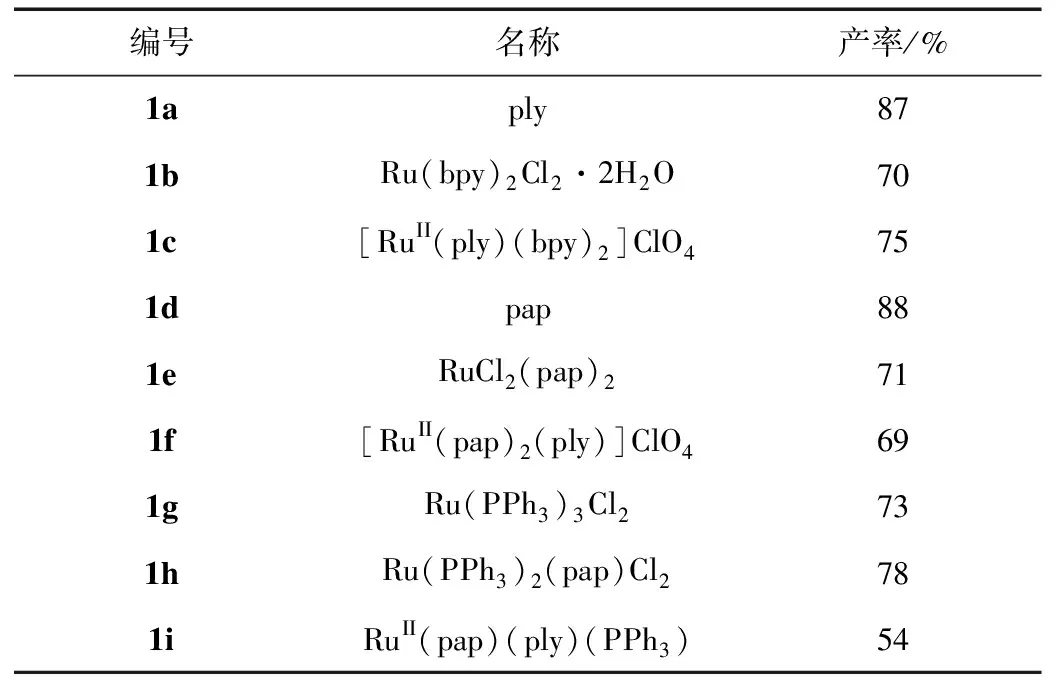
表1 9-氧化邻苯二酚和钌配合物合成的产率
由表1可知,这9-氧化邻苯二酚和钌的三种配合物的每一步合成均有中等至良好的产率。
1a:Isolation by column chromatography (PE/EtOAc: 10/1) as a yellow oil.1H NMR (400 MHz, CDCl3):δ ppm 16.06 (d, 1H, OH), 8.13~8.10 (m, 2H, ArH), 8.05~8.03 (m, 2H, ArH), 7.63~7.59 (m, 1H, ArH), 1.57 (m, 2H, ArH)。
1b:Isolation by column chromatography (CH2Cl2/MeCN: 5/1) as a red-brown oil.1H NMR (400 MHz, CDCl3):δ ppm 8.70~8.65 (m, 4H, ArH), 8.42~8.38 (m, 4H, ArH), 7.82~7.77 (m, 4H, ArH), 7.30~7.26 (m, 4H, ArH)。
1c:Isolation by column chromatography (CH2Cl2/MeCN: 5/1) as a purple oil.1H NMR (400 MHz, DMSO):δ ppm 8.85~8.79 (m, 2H, ArH), 8.74~8.69 (m, 2H, ArH), 8.68~8.64 (m, 2H, ArH), 8.21~8.12 (m, 2H, ArH), 8.08~8.02 (m, 2H, ArH), 7.96~7.89 (m, 4H, ArH), 7.82~7.77 (m, 2H, ArH), 7.70~7.61 (m, 2H, ArH), 7.47~7.39 (m, 1H, ArH), 7.32~7.24 (m, 2H, ArH), 6.92~6.86 (m, 2H, ArH)。
1d:Isolation by column chromatography (CH2Cl2/MeCN: 5/1) as yellow oil.1H NMR (400 MHz, CDCl3): δ ppm 8.79~8.71 (m, 1H, ArH), 8.10~8.02 (m, 2H, ArH), 7.95~7.88 (m, 1H, ArH), 7.86~7.81 (m, 1H, ArH), 7.58~7.49 (m, 3H, ArH), 7.44~7.38 (m, 1H, ArH)。
1e:Isolation by column chromatography (CH2Cl2/MeCN: 5/1) as a purple oil.1H NMR (400 MHz, CDCl3): δ ppm 8.96~8.89 (m, 2H, ArH), 8.63~8.58 (m, 2H, ArH), 8.23~8.16 (m, 2H, ArH), 7.85~7.79 (m, 1H, ArH), 7.57~7.50 (m, 4H, ArH), 7.21~7.15 (m, 2H, ArH), 7.04~6.92 (m, 4H, ArH)。
1f:Isolation by column chromatography (CH2Cl2/MeCN: 5/1) as a purple oil.1H NMR (400 MHz, DMSO): δ ppm 9.13~9.05 (m, 1H, ArH), 9.02~8.97 (m, 1H, ArH), 8.51~8.42 (m, 2H, ArH), 8.38~8.34 (m, 1H, ArH), 8.19~7.97 (m, 5H, ArH), 7.94~7.89 (m, 1H, ArH), 7.88~7.82 (m, 1H, ArH), 7.75~7.66 (m, 1H, ArH), 7.56~7.51 (m, 1H, ArH), 7.45~7.33 (m, 5H, ArH), 7.17~7.07 (m, 3H, ArH), 6.90~6.80 (m, 3H, ArH), 5.77~5.73 (m, 2H, ArH)。
1g:Isolation by column chromatography (MeOH) as a brown oil.1H NMR (400 MHz, CDCl3): δ ppm 7.38~7.37 (m, 27H, ArH), 7.37~7.36 (m, 18H, ArH)。
1h:Isolation by column chromatography (C6H14) as a fuchsia oil.1H NMR (400 MHz, CDCl3): δ ppm 7.44~7.39 (m, 15H, ArH), 7.15~7.09 (m, 8H, ArH), 7.04~6.98 (m, 15H, ArH)。
1i:Isolation by column chromatography (CH2Cl2/MeCN: 5/1) as a purple oil.1H NMR (400 MHz, DMSO):δ ppm 8.07~7.94 (m, 4H, ArH), 7.69~7.54 (m, 12H, ArH), 7.30~7.18 (m, 4H, ArH), 7.14~7.02 (m, 6H, ArH), 6.85~6.73 (m, 5H, ArH), 3.24~2.96 (m, 2H, ArH)。
2.2 催化苄基衍生物的氧化
我们根据一定的反应条件对一定数量的苄基衍生物进行了选择性氧化反应,其氧化反应结果如表2所示。
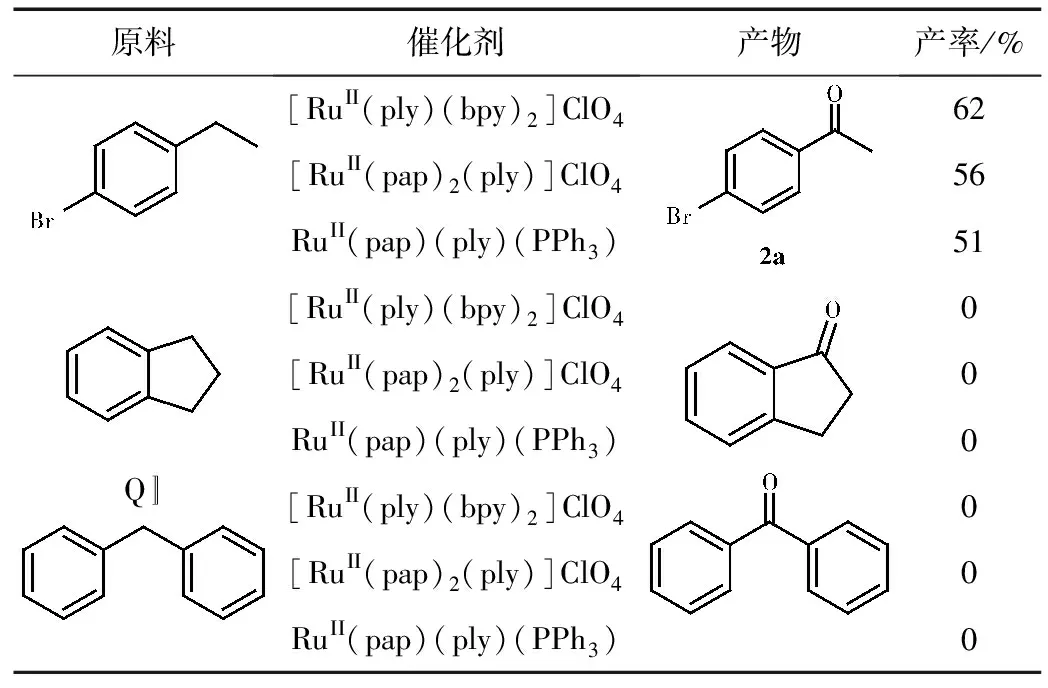
表2 催化苄基衍生物氧化的实验结果
由表2可知,这三种催化剂在催化苄基衍生物C-H键到羰基效果较差,仅有一种苄基衍生物有中等产率。
2a:Isolation by column chromatography (PE/EtOAc: 10/1) as a colorless oil.1H NMR (400 MHz, CDCl3): δ ppm 7.82 (d, 2H,J=8 Hz, ArH), 7.61 (d, 2H,J=8 Hz, ArH), 2.59 (s, 3H, CH3)。
2.3 催化胺的氧化
我们根据一定的反应条件对一定数量的胺进行了选择性氧化反应,其氧化反应结果如表3所示。
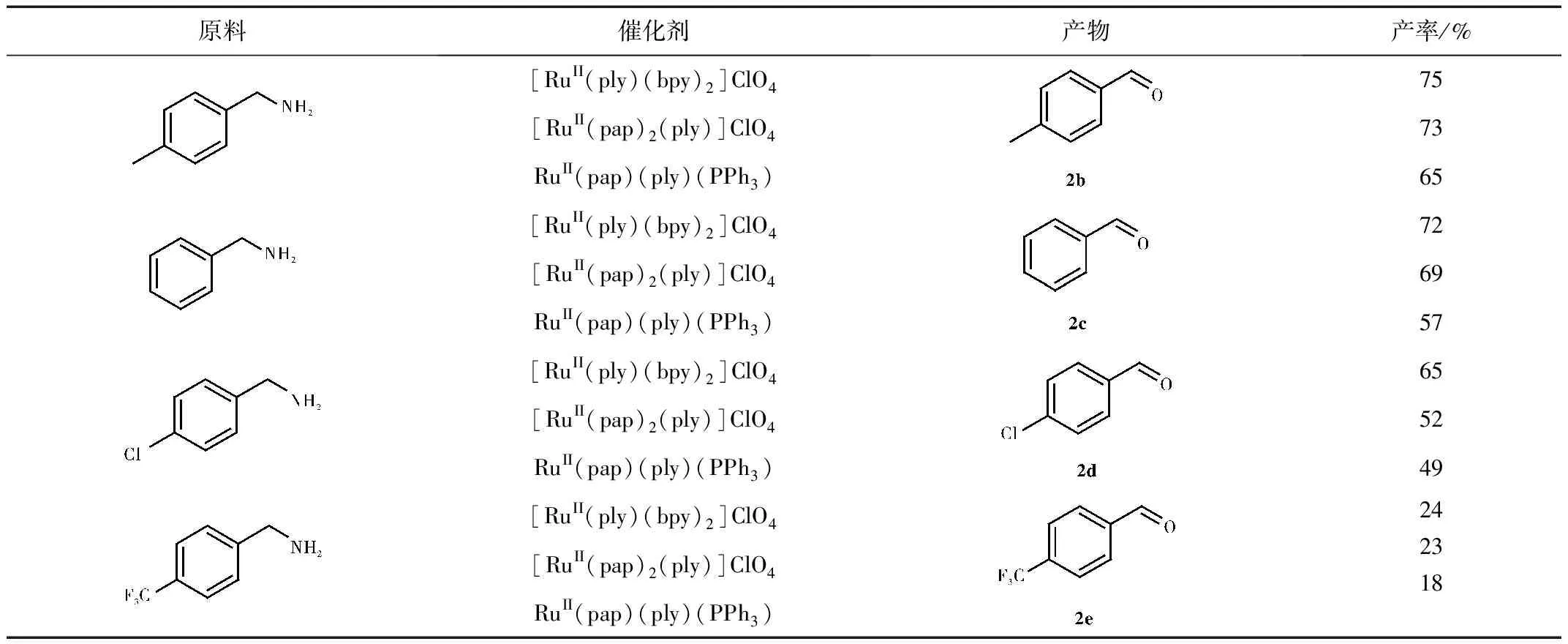
表3 催化胺氧化的实验结果
由表3可知,这三种催化剂在催化氧化胺类到醛类效果较好,75%的胺类有中等产率,且总体看来催化效果[RuII(ply)(bpy)2]ClO4最好,[RuII(pap)2(ply)]ClO4次之,RuII(pap)(ply)(PPh3)最末。
2.4 催化硫化物的氧化
我们根据一定的反应条件对一定数量的硫化物进行了选择性氧化反应,其氧化反应结果如表4所示。
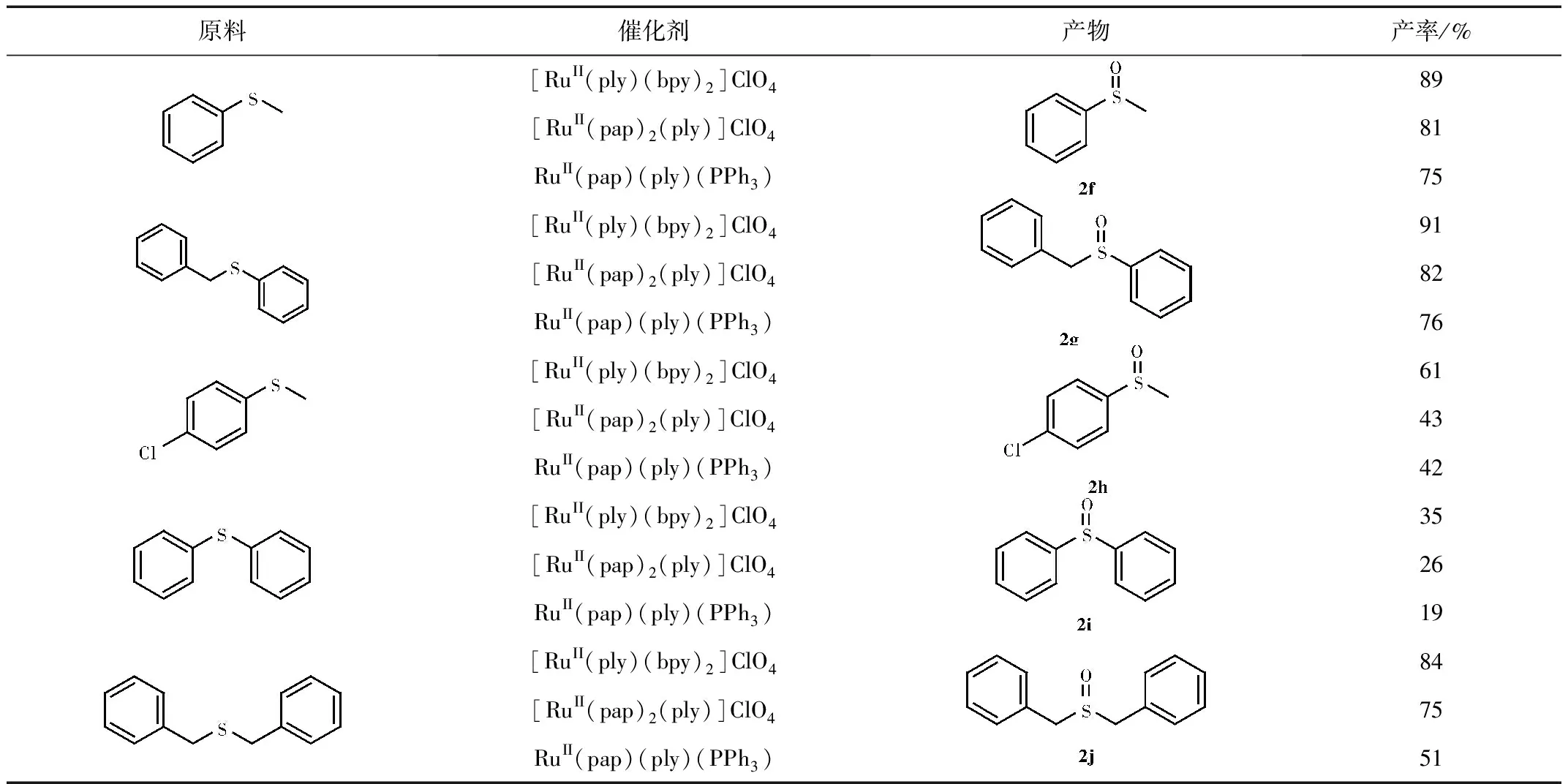
表4 催化硫化物氧化的实验结果
由表4可知,这三种催化剂在催化氧化硫化物到亚砜效果较好,每种硫化物都获得了中等至良好的产率,且整体来看催化效果[RuII(ply)(bpy)2]ClO4最好,[RuII(pap)2(ply)]ClO4次之,RuII(pap)(ply)(PPh3)最末。
2f:Isolation by column chromatography (PE/EtOAc: 5/1) as a white viscous oil.1H NMR (400 MHz, CDCl3):δ ppm 7.67~7.64 (m, 2H, ArH), 7.56~7.48 (m, 3H, ArH), 2.73 (s, 3H, CH3)。
2g:Isolation by column chromatography (PE/EtOAc: 5/1) as a white viscous oil.1H NMR (400 MHz, CDCl3): δ ppm 7.47~7.36 (m, 5H, ArH), 7.29~7.21 (m, 3H, ArH), 6.98~6.96 (m, 2H, ArH), 4.00~3.97 (m, 2H, CH2)。
2h:Isolation by column chromatography (PE/EtOAc: 5/1) as a white viscous oil.1H NMR (400 MHz, CDCl3): δ ppm 7.6 (d, 2H,J=8 Hz, ArH), 7.51 (d, 2H,J=8 Hz, ArH), 2.73 (s, 3H, CH3)。
2i:Isolation by column chromatography (PE/EtOAc: 5/1) as a white viscous oil.1H NMR (400 MHz, CDCl3): δ ppm 7.66~7.64 (m, 4H, ArH), 7.49~7.42 (m, 6H, ArH)。
2j:Isolation by column chromatography (PE/EtOAc: 5/1) as a white viscous oil.1H NMR (400 MHz, CDCl3):δ ppm 7.40~7.34 (m, 3H, ArH), 7.30~7.28 (m, 2H, ArH), 3.94~3.86(m, 2H, CH2)。
3 结 论
9-氧化邻苯二酚与钌制备了三种新型配合物光催化剂,制备过程的每一步均有中等至良好的收率。9-氧化邻苯二酚与钌结合的三种配合物在氧化胺类到醛类和氧化硫化物到亚砜都有中等的产率,其中整体看来催化效果[RuII(ply)(bpy)2]ClO4最好,[RuII(pap)2(ply)]ClO4次之,RuII(pap)(ply)(PPh3)最末,但是这三种催化剂在苄基衍生物C-H键到羰基效果较差,后续需继续摸索条件。
猜你喜欢
净水技术(2022年4期)2022-04-12
化学分析计量(2021年6期)2021-06-22
闽南师范大学学报(自然科学版)(2020年1期)2020-06-05
生物技术通报(2019年9期)2019-09-18
百科知识(2016年18期)2016-10-28
中国洗涤用品工业(2015年9期)2015-02-28
中国塑料(2014年10期)2014-10-17
天然产物研究与开发(2014年6期)2014-04-27
郑州大学学报(理学版)(2014年4期)2014-03-01
郑州大学学报(工学版)(2012年1期)2012-09-13
